
Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.

Thrombotic thrombocytopenic purpura (TTP) is a blood disorder that results in blood clots forming in small blood vessels throughout the body. This results in a low platelet count, low red blood cells due to their breakdown, and often kidney, heart, and brain dysfunction. Symptoms may include large bruises, fever, weakness, shortness of breath, confusion, and headache. Repeated episodes may occur.

Uremia is the term for high levels of urea in the blood. Urea is one of the primary components of urine. It can be defined as an excess in the blood of amino acid and protein metabolism end products, such as urea and creatinine, which would be normally excreted in the urine. Uremic syndrome can be defined as the terminal clinical manifestation of kidney failure. It is the signs, symptoms and results from laboratory tests which result from inadequate excretory, regulatory, and endocrine function of the kidneys. Both uremia and uremic syndrome have been used interchangeably to denote a very high plasma urea concentration that is the result of renal failure. The former denotation will be used for the rest of the article.
Microangiopathic hemolytic anemia (MAHA) is a microangiopathic subgroup of hemolytic anemia caused by factors in the small blood vessels. It is identified by the finding of anemia and schistocytes on microscopy of the blood film.
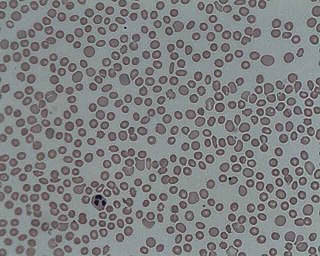
In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.

Hemolytic–uremic syndrome (HUS) is a group of blood disorders characterized by low red blood cells, acute kidney failure, and low platelets. Initial symptoms typically include bloody diarrhea, fever, vomiting, and weakness. Kidney problems and low platelets then occur as the diarrhea progresses. Children are more commonly affected, but most children recover without permanent damage to their health, although some children may have serious and sometimes life-threatening complications. Adults, especially the elderly, may present a more complicated presentation. Complications may include neurological problems and heart failure.

Von Willebrand factor (VWF) is a blood glycoprotein involved in hemostasis, specifically, platelet adhesion. It is deficient and/or defective in von Willebrand disease and is involved in many other diseases, including thrombotic thrombocytopenic purpura, Heyde's syndrome, and possibly hemolytic–uremic syndrome. Increased plasma levels in many cardiovascular, neoplastic, metabolic, and connective tissue diseases are presumed to arise from adverse changes to the endothelium, and may predict an increased risk of thrombosis.
Hemoglobinuria is a condition in which the oxygen transport protein hemoglobin is found in abnormally high concentrations in the urine. The condition is caused by excessive intravascular hemolysis, in which large numbers of red blood cells (RBCs) are destroyed, thereby releasing free hemoglobin into the plasma. Excess hemoglobin is filtered by the kidneys, which excrete it into the urine, giving urine a purple color. Hemoglobinuria can lead to acute tubular necrosis which is an uncommon cause of a death of uni-traumatic patients recovering in the ICU.

Plasmapheresis is the removal, treatment, and return or exchange of blood plasma or components thereof from and to the blood circulation. It is thus an extracorporeal therapy, a medical procedure performed outside the body.

A schistocyte or schizocyte is a fragmented part of a red blood cell. Schistocytes are typically irregularly shaped, jagged, and have two pointed ends.

Hepatorenal syndrome is a life-threatening medical condition that consists of rapid deterioration in kidney function in individuals with cirrhosis or fulminant liver failure. HRS is usually fatal unless a liver transplant is performed, although various treatments, such as dialysis, can prevent advancement of the condition.

Nephritic syndrome is a syndrome comprising signs of nephritis, which is kidney disease involving inflammation. It often occurs in the glomerulus, where it is called glomerulonephritis. Glomerulonephritis is characterized by inflammation and thinning of the glomerular basement membrane and the occurrence of small pores in the podocytes of the glomerulus. These pores become large enough to permit both proteins and red blood cells to pass into the urine. By contrast, nephrotic syndrome is characterized by proteinuria and a constellation of other symptoms that specifically do not include hematuria. Nephritic syndrome, like nephrotic syndrome, may involve low level of albumin in the blood due to the protein albumin moving from the blood to the urine.
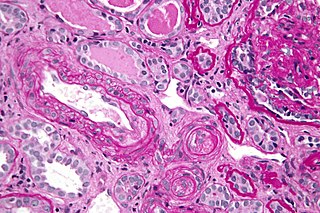
Thrombotic microangiopathy (TMA) is a pathology that results in thrombosis in capillaries and arterioles, due to an endothelial injury. It may be seen in association with thrombocytopenia, anemia, purpura and kidney failure.

Calciphylaxis, also known as calcific uremic arteriolopathy (CUA) or “Grey Scale”, is a rare syndrome characterized by painful skin lesions. The pathogenesis of calciphylaxis is unclear but believed to involve calcification of the small blood vessels located within the fatty tissue and deeper layers of the skin, blood clots, and eventual death of skin cells due to lack of blood flow. It is seen mostly in people with end-stage kidney disease but can occur in the earlier stages of chronic kidney disease and rarely in people with normally functioning kidneys. Calciphylaxis is a rare but serious disease, believed to affect 1-4% of all dialysis patients. It results in chronic non-healing wounds and indicates poor prognosis, with typical life expectancy of less than one year.

Eculizumab, sold under the brand name Soliris among others, is a recombinant humanized monoclonal antibody used to treat paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), generalized myasthenia gravis, and neuromyelitis optica. In people with PNH, it reduces both the destruction of red blood cells and need for blood transfusion, but does not appear to affect the risk of death. Eculizumab was the first drug approved for each of its uses, and its approval was granted based on small trials. It is given by intravenous infusion.

Complement factor I, also known as C3b/C4b inactivator, is a protein that in humans is encoded by the CFI gene. Complement factor I is a protein of the complement system, first isolated in 1966 in guinea pig serum, that regulates complement activation by cleaving cell-bound or fluid phase C3b and C4b. It is a soluble glycoprotein that circulates in human blood at an average concentration of 35 μg/mL.

C3b is the larger of two elements formed by the cleavage of complement component 3, and is considered an important part of the innate immune system. C3b is potent in opsonization: tagging pathogens, immune complexes (antigen-antibody), and apoptotic cells for phagocytosis. Additionally, C3b plays a role in forming a C3 convertase when bound to Factor B, or a C5 convertase when bound to C4b and C2b or when an additional C3b molecule binds to the C3bBb complex.
Onconephrology is a specialty in nephrology that deals with the study of kidney diseases in cancer patients. A nephrologist who takes care of patients with cancer and kidney disease is called an onconephrologist. This branch of nephrology encompasses nephrotoxicity associated with existing and novel chemotherapeutics, kidney disease as it pertains to stem cell transplant, paraneoplastic kidney disorders, paraproteinemias, electrolyte disorders associated with cancer, and more as discussed below.
Upshaw–Schulman syndrome (USS) is the recessively inherited form of thrombotic thrombocytopenic purpura (TTP), a rare and complex blood coagulation disease. USS is caused by the absence of the ADAMTS13 protease resulting in the persistence of ultra large von Willebrand factor multimers (ULVWF), causing episodes of acute thrombotic microangiopathy with disseminated multiple small vessel obstructions. These obstructions deprive downstream tissues from blood and oxygen, which can result in tissue damage and death. The presentation of an acute USS episode is variable but usually associated with thrombocytopenia, microangiopathic hemolytic anemia (MAHA) with schistocytes on the peripheral blood smear, fever and signs of ischemic organ damage in the brain, kidney and heart.
Ravulizumab, sold under the brand name Ultomiris, is a humanized monoclonal antibody complement inhibitor medication designed for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome. It is designed to bind to and prevent the activation of Complement component 5 (C5).