
Marfan syndrome (MFS) is a multi-systemic genetic disorder that affects the connective tissue. Those with the condition tend to be tall and thin, with long arms, legs, fingers, and toes. They also typically have exceptionally flexible joints and abnormally curved spines. The most serious complications involve the heart and aorta, with an increased risk of mitral valve prolapse and aortic aneurysm. The lungs, eyes, bones, and the covering of the spinal cord are also commonly affected. The severity of the symptoms is variable.

Arthrogryposis (AMC) describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning 'curving of joints'.

Arachnodactyly is a medical condition that is characterized by fingers and toes that are abnormally long and slender, in comparison to the palm of the hand and arch of the foot. In some cases, the thumbs of an individual with the condition are pulled inwards towards the palm. This condition is present at birth.

Fibrillin is a glycoprotein, which is essential for the formation of elastic fibers found in connective tissue. Fibrillin is secreted into the extracellular matrix by fibroblasts and becomes incorporated into the insoluble microfibrils, which appear to provide a scaffold for deposition of elastin.
A connective tissue disease (collagenosis) is any disease that has the connective tissues of the body as a target of pathology. Connective tissue is any type of biological tissue with an extensive extracellular matrix that supports, binds together, and protects organs. These tissues form a framework, or matrix, for the body, and are composed of two major structural protein molecules: collagen and elastin. There are many different types of collagen protein in each of the body's tissues. Elastin has the capability of stretching and returning to its original length—like a spring or rubber band. Elastin is the major component of ligaments and skin. In patients with connective tissue disease, it is common for collagen and elastin to become injured by inflammation (ICT). Many connective tissue diseases feature abnormal immune system activity with inflammation in tissues as a result of an immune system that is directed against one's own body tissues (autoimmunity).

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.
Acromicric dysplasia is an extremely rare inherited disorder characterized by abnormally short hands and feet, growth retardation and delayed bone maturation leading to short stature. Most cases have occurred randomly for no apparent reason (sporadically). However, autosomal dominant inheritance has not been ruled out.

Loeys–Dietz syndrome (LDS) is an autosomal dominant genetic connective tissue disorder. It has features similar to Marfan syndrome and Ehlers–Danlos syndrome. The disorder is marked by aneurysms in the aorta, often in children, and the aorta may also undergo sudden dissection in the weakened layers of the wall of the aorta. Aneurysms and dissections also can occur in arteries other than the aorta. Because aneurysms in children tend to rupture early, children are at greater risk for dying if the syndrome is not identified. Surgery to repair aortic aneurysms is essential for treatment.
MASS syndrome is a medical disorder of the connective tissue similar to Marfan syndrome. MASS stands for mitral valve prolapse, aortic root diameter at upper limits of normal for body size, stretch marks of the skin, and skeletal conditions similar to Marfan syndrome. It is caused by a mutation in the FBN1 gene, which encodes fibrillin-1. Fibrillin-1 is an extracellular matrix protein that is found in microfibrils; defects in the fibrillin-1 protein cause the malfunctioning of microfibrils, which results in improper stretching of ligaments, blood vessels, and skin.

Arterial tortuosity syndrome is an extremely rare congenital connective tissue condition disorder characterized by tortuosity, elongation, stenosis, or aneurysms in major and medium-size arteries including the aorta. It is associated with hyperextensible skin and hypermobility of joints, however symptoms vary depending on the person. Because ATS is so rare, relatively little is known about the disease compared to more common diseases.

Fibrillin-1 is a protein that in humans is encoded by the FBN1 gene, located on chromosome 15. It is a large, extracellular matrix glycoprotein that serves as a structural component of 10-12 nm calcium-binding microfibrils. These microfibrils provide force bearing structural support in elastic and nonelastic connective tissue throughout the body. Mutations altering the protein can result in a variety of phenotypic effects differing widely in their severity, including fetal death, developmental problems, Marfan syndrome or in some cases Weill-Marchesani syndrome.
Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.
Haim–Munk syndrome is a skin disease caused, like Papillon-Lefevre Syndrome, by a mutation in the cathepsin C gene. One of its features is thick curved finger and toenails.
Setleis syndrome is a very rare genetic condition characterized by facial skin abnormalities and double upper eyelashes and missing lower eyelashes. It belongs to a group of diseases known as ectodermal dysplasias. Ectodermal dysplasias typically affect the hair, teeth, nails, and/or skin. Setleis syndrome is characterized by distinctive abnormalities of the facial area that may be apparent at birth (congenital). Most affected infants have multiple, scar-like, circular depressions on both temples (bitemporal). These marks closely resemble those made when forceps are used to assist delivery. The range and severity of symptoms may vary from case to case.
Stiff skin syndrome is a cutaneous condition characterized by ‘rock hard’ induration, thickening of the skin and subcutaneous tissues, limited joint mobility, and mild hypertrichosis in infancy or early childhood. Immunologic abnormalities or vascular hyperactivity are not present in patients.
Shprintzen–Goldberg syndrome is a congenital multiple-anomaly syndrome that has craniosynostosis, multiple abdominal hernias, cognitive impairment, and other skeletal malformations as key features. Several reports have linked the syndrome to a mutation in the FBN1 gene, but these cases do not resemble those initially described in the medical literature in 1982 by Shprintzen and Goldberg, and Greally et al. in 1998 failed to find a causal link to FBN1. At this time, the cause of Shprintzen–Goldberg syndrome has been identified as a mutation in the gene SKI located on chromosome 1 at the p36 locus. The syndrome is rare with fewer than 50 cases described in the medical literature to date.
Myhre syndrome is a rare genetic disorder inherited in an autosomal dominant fashion. It is caused by mutation in SMAD4 gene.
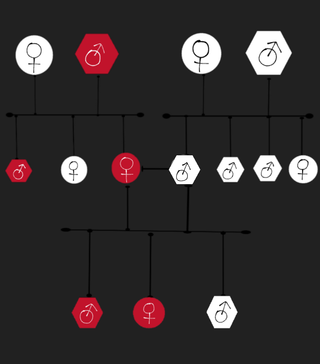
Familial thoracic aortic aneurysm and aortic dissection is a very rare vascular genetic disorder, it's characterized by recurrent thoracic aortic aneurysms and aortic dissections within a family, these mentioned complications affect one or more aortic segments without any other disease being associated with them. People with this disorder have a higher chance of having a potentially fatal aortic rupture. This disorder is the cause of 20% of thoracic aortic aneurysms
Cryptorchidism-arachnodactyly-intellectual disability syndrome is a rare multi-systemic genetic disorder of unknown prevalence which is characterized by psycho-motor developmental delay, severe intellectual disabilities, severe muscle hypoplasia, absence of subcutaneous fat, generalized contractures, dolichocephaly, esotropia, asymmetric ears, and high palate, kyphoscoliosis, unilateral hypoplasia of the bronchial system, recurrent respiratory tract infections, atelectasis, arachnodactyly, cryptorchidism, hypospadias, and testicular agenesis. No new cases have been reported since 1970.
