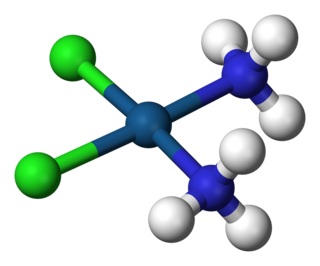
In coordination chemistry, a ligand is an ion or molecule that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".

An excimer is a short-lived dimeric or heterodimeric molecule formed from two species, at least one of which has completely filled valence shell by electrons. In this case, formation of molecules is possible only if such atom is in an electronic excited state. Heteronuclear molecules and molecules that have more than two species are also called exciplex molecules. Excimers are often diatomic and are composed of two atoms or molecules that would not bond if both were in the ground state. The lifetime of an excimer is very short, on the order of nanoseconds. Binding of a larger number of excited atoms form Rydberg matter clusters, the lifetime of which can exceed many seconds.
The color of chemicals is a physical property of chemicals that in most cases comes from the excitation of electrons due to an absorption of energy performed by the chemical. What is seen by the eye is not the color absorbed, but the complementary color from the removal of the absorbed wavelengths. This spectral perspective was first noted in atomic spectroscopy.
In chemistry, Molecular orbital (MO) theory is a method for describing the electronic structure of molecules using quantum mechanics. Electrons are not assigned to individual bonds between atoms, but are treated as moving under the influence of the nuclei in the whole molecule. The spatial and energetic properties of electrons are described by quantum mechanics as molecular orbitals surround two or more atoms in a molecule and contain valence electrons between atoms. Molecular orbital theory, which was proposed in the early twentieth century, revolutionized the study of bonding by approximating the states of bonded electrons—the molecular orbitals—as linear combinations of atomic orbitals (LCAO). These approximations are now made by applying the density functional theory (DFT) or Hartree–Fock (HF) models to the Schrödinger equation.

Kenichi Fukui was a Japanese chemist, known as the first Asian scientist to receive a chemistry Nobel Prize.
Crystal Field Theory (CFT) is a model that describes the breaking of degeneracies of electron orbital states, usually d or f orbitals, due to a static electric field produced by a surrounding charge distribution. This theory has been used to describe various spectroscopies of transition metal coordination complexes, in particular optical spectra (colors). CFT successfully accounts for some magnetic properties, colors, hydration enthalpies, and spinel structures of transition metal complexes, but it does not attempt to describe bonding. CFT was developed by physicists Hans Bethe and John Hasbrouck van Vleck in the 1930s. CFT was subsequently combined with molecular orbital theory to form the more realistic and complex ligand field theory (LFT), which delivers insight into the process of chemical bonding in transition metal complexes.
A charge-transfer complex or electron-donor-acceptor complex is an association of two or more molecules, or of different parts of one large molecule, in which a fraction of electronic charge is transferred between the molecular entities. The resulting electrostatic attraction provides a stabilizing force for the molecular complex. The source molecule from which the charge is transferred is called the electron donor and the receiving species is called the electron acceptor.
Ligand field theory (LFT) describes the bonding, orbital arrangement, and other characteristics of coordination complexes. It represents an application of molecular orbital theory to transition metal complexes. A transition metal ion has nine valence atomic orbitals - consisting of five nd, three (n+1)p, and one (n+1)s orbitals. These orbitals are of appropriate energy to form bonding interaction with ligands. The LFT analysis is highly dependent on the geometry of the complex, but most explanations begin by describing octahedral complexes, where six ligands coordinate to the metal. Other complexes can be described by reference to crystal field theory.

The isolobal principle is a strategy used in organometallic chemistry to relate the structure of organic and inorganic molecular fragments in order to predict bonding properties of organometallic compounds. Roald Hoffmann described molecular fragments as isolobal "if the number, symmetry properties, approximate energy and shape of the frontier orbitals and the number of electrons in them are similar – not identical, but similar." One can predict the bonding and reactivity of a lesser-known species from that of a better-known species if the two molecular fragments have similar frontier orbitals, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Isolobal compounds are analogues to isoelectronic compounds that share the same number of valence electrons and structure. A graphic representation of isolobal structures, with the isolobal pairs connected through a double-headed arrow with half an orbital below, is found in Figure 1.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives.
An electrocyclic reaction can either be classified as conrotatory or disrotatory based on the rotation at each end of the molecule. In conrotatory mode, both atomic orbitals of the end groups turn in the same direction. In disrotatory mode, the atomic orbitals of the end groups turn in opposite directions. The cis/trans geometry of the final product is directly decided by the difference between conrotation and disrotation.
Koopmans' theorem states that in closed-shell Hartree–Fock theory (HF), the first ionization energy of a molecular system is equal to the negative of the orbital energy of the highest occupied molecular orbital (HOMO). This theorem is named after Tjalling Koopmans, who published this result in 1934.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.
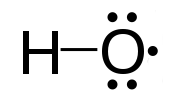
A non-bonding orbital, also known as non-bonding molecular orbital (NBMO), is a molecular orbital whose occupation by electrons neither increases nor decreases the bond order between the involved atoms. Non-bonding orbitals are often designated by the letter n in molecular orbital diagrams and electron transition notations. Non-bonding orbitals are the equivalent in molecular orbital theory of the lone pairs in Lewis structures. The energy level of a non-bonding orbital is typically in between the lower energy of a valence shell bonding orbital and the higher energy of a corresponding antibonding orbital. As such, a non-bonding orbital with electrons would commonly be a HOMO.
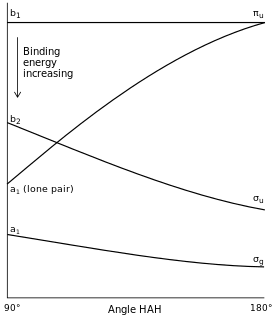
Walsh diagrams, often called angular coordinate diagrams or correlation diagrams, are representations of calculated orbital binding energies of a molecule versus a distortion coordinate, used for making quick predictions about the geometries of small molecules. By plotting the change in molecular orbital levels of a molecule as a function of geometrical change, Walsh diagrams explain why molecules are more stable in certain spatial configurations.
The Inverse electron demand Diels–Alder reaction, or DAINV or IEDDA is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene. During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
In computational chemistry, the Fukui function or frontier function is a function that describes the electron density in a frontier orbital, as a result of a small change in the total number of electrons. The condensed Fukui function or condensed reactivity indicator is the same idea, but applied to an atom within a molecule, rather than a point in three-dimensional space.
A metal-centered cycloaddition is a subtype of the more general class of cycloaddition reactions. In such reactions "two or more unsaturated molecules unite directly to form a ring", incorporating a metal bonded to one or more of the molecules. Cycloadditions involving metal centers are a staple of organic and organometallic chemistry, and are involved in many industrially-valuable synthetic processes.