A haplotype is a group of alleles in an organism that are inherited together from a single parent. However, there are other uses of this term. First, it is used to mean a collection of specific alleles in a cluster of tightly linked genes on a chromosome that are likely to be inherited together—that is, they are likely to be conserved as a sequence that survives the descent of many generations of reproduction. A second use is to mean a set of linked single-nucleotide polymorphism (SNP) alleles that tend to always occur together. It is thought that identifying these statistical associations and few alleles of a specific haplotype sequence can facilitate identifying all other such polymorphic sites that are nearby on the chromosome. Such information is critical for investigating the genetics of common diseases; which in fact have been investigated in humans by the International HapMap Project. Thirdly, many human genetic testing companies use the term in a third way: to refer to an individual collection of specific mutations within a given genetic segment;.
A quantitative trait locus (QTL) is a locus which correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying and sequencing the actual genes that cause the trait variation.
Genetics, a discipline of biology, is the science of heredity and variation in living organisms.
Y linkage, also known as sex linkage or holandric inheritance describes traits that are produced by genes located on the Y chromosome.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.
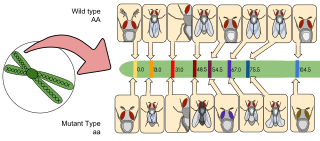
Gene maps help describe the spatial arrangement of genes on a chromosome. Genes are designated to a specific location on a chromosome known as the locus and can be used as molecular markers to find the distance between other genes on a chromosome. Maps provide researchers with the opportunity to predict the inheritance patterns of specific traits, which can eventually lead to a better understanding of disease-linked traits.
A molecular marker is a molecule contained within a sample taken from an organism or other matter. It can be used to reveal certain characteristics about the respective source. DNA, for example, is a molecular marker containing information about genetic disorders, genealogy and the evolutionary history of life. Specific regions of the DNA are used for diagnosing the autosomal recessive genetic disorder cystic fibrosis, taxonomic affinity (phylogenetics) and identity. Further, life forms are known to shed unique chemicals, including DNA, into the environment as evidence of their presence in a particular location. Other biological markers, like proteins, are used in diagnostic tests for complex neurodegenerative disorders, such as Alzheimer's disease. Non-biological molecular markers are also used, for example, in environmental studies.
Neil Risch is an American human geneticist and professor at the University of California, San Francisco (UCSF). Risch is the Lamond Family Foundation Distinguished Professor in Human Genetics and Director of the Institute for Human Genetics and Professor of Epidemiology and Biostatistics at UCSF.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.
Xq28 is a chromosome band and genetic marker situated at the tip of the X chromosome which has been studied since at least 1980. The band contains three distinct regions, totaling about 8 Mbp of genetic information. The marker came to the public eye in 1993 when studies by Dean Hamer and others indicated a link between the Xq28 marker and male sexual orientation.
In genetics, complete linkage is defined as the state in which two loci are so close together that alleles of these loci are virtually never separated by crossing over. The closer the physical location of two genes on the DNA, the less likely they are to be separated by a crossing-over event. In the case of male Drosophila there is complete absence of recombinant types due to absence of crossing over. This means that all of the genes that start out on a single chromosome, will end up on that same chromosome in their original configuration. In the absence of recombination, only parental phenotypes are expected.
The Elston–Stewart algorithm is an algorithm for computing the likelihood of observed genotype data given a pedigree. It is due to Robert Elston and John Stewart. It can handle relatively large pedigrees providing they are (almost) outbred. Its computation time is exponential in the number of markers. It is used in the analysis of genetic linkage.
Dr. Robert C. Elston is a statistical geneticist and distinguished professor emeritus at Case Western Reserve University. He was born in London, England. He is one of the eponyms of the Elston–Stewart algorithm.
Association mapping (genetics), also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Quantitative trait loci mapping or QTL mapping is the process of identifying genomic regions that potentially contain genes responsible for important economic, health or environmental characters. Mapping QTLs is an important activity that plant breeders and geneticists routinely use to associate potential causal genes with phenotypes of interest. Family-based QTL mapping is a variant of QTL mapping where multiple-families are used.
Philip Palmer Green is a theoretical and computational biologist noted for developing important algorithms and procedures used in Gene mapping and DNA sequencing. He earned his doctorate from Berkeley in mathematics in 1976 with a dissertation on C*-algebra under the direction of Marc Rieffel, but transitioned from pure mathematics into applied work in biology and bioinformatics. Green has obtained numerous important results, including in developing Phred, a widely used DNA trace analyzer, in mapping techniques, and in genetic analysis. Green was elected to the National Academy of Sciences in 2001 and won the Gairdner Award in 2002.
Mega2 allows the applied statistical geneticist to convert one's data from several input formats to a large number output formats suitable for analysis by commonly used software packages. In a typical human genetics study, the analyst often needs to use a variety of different software programs to analyze the data, and these programs usually require that the data be formatted to their precise input specifications. Conversion of one's data into these multiple different formats can be tedious, time-consuming, and error-prone. Mega2, by providing validated conversion pipelines, can accelerate the analyses while reducing errors.
Dr. Rohan L. Fernando is a Professor of Quantitative Genetics in the Department of Animal Science at Iowa State University (ISU), USA. Although recognized for his work in many facets of genetics, Dr. Fernando’s efforts have focused primarily on theory and methods for use of genetic markers in breeding, theory and methods for genetic evaluations of crossbred animals, methodology related to the estimation of genetic parameters and the prediction of genetic merit in populations undergoing selection and non-random mating, bayesian methodology for analysis of unbalanced mixed model data, optimization of breeding programs, and use of computer simulation to study dynamics of genetic system.