A neuron, neurone, or nerve cell is an electrically excitable cell that communicates with other cells via synapses - specialized connections that commonly use minute amounts of neurotransmitters to pass the electric signal from the presynaptic neuron to the target cell through the synaptic gap. The neuron is the main component of nervous tissue in all animals except sponges and placozoa. Non-animals like plants and fungi do not have nerve cells.
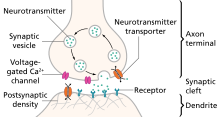
A neurotransmitter receptor is a membrane receptor protein that is activated by a neurotransmitter. Chemicals on the outside of the cell, such as a neurotransmitter, can bump into the cell's membrane, in which there are receptors. If a neurotransmitter bumps into its corresponding receptor, they will bind and can trigger other events to occur inside the cell. Therefore, a membrane receptor is part of the molecular machinery that allows cells to communicate with one another. A neurotransmitter receptor is a class of receptors that specifically binds with neurotransmitters as opposed to other molecules.

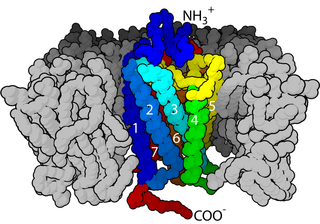
The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a “coincidence detector” and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.
In neuroscience, synaptic plasticity is the ability of synapses to strengthen or weaken over time, in response to increases or decreases in their activity. Since memories are postulated to be represented by vastly interconnected neural circuits in the brain, synaptic plasticity is one of the important neurochemical foundations of learning and memory.
An inhibitory postsynaptic potential (IPSP) is a kind of synaptic potential that makes a postsynaptic neuron less likely to generate an action potential. IPSP were first investigated in motorneurons by David P. C. Lloyd, John Eccles and Rodolfo Llinás in the 1950s and 1960s. The opposite of an inhibitory postsynaptic potential is an excitatory postsynaptic potential (EPSP), which is a synaptic potential that makes a postsynaptic neuron more likely to generate an action potential. IPSPs can take place at all chemical synapses, which use the secretion of neurotransmitters to create cell to cell signalling. Inhibitory presynaptic neurons release neurotransmitters that then bind to the postsynaptic receptors; this induces a change in the permeability of the postsynaptic neuronal membrane to particular ions. An electric current that changes the postsynaptic membrane potential to create a more negative postsynaptic potential is generated, i.e. the postsynaptic membrane potential becomes more negative than the resting membrane potential, and this is called hyperpolarisation. To generate an action potential, the postsynaptic membrane must depolarize—the membrane potential must reach a voltage threshold more positive than the resting membrane potential. Therefore, hyperpolarisation of the postsynaptic membrane makes it less likely for depolarisation to sufficiently occur to generate an action potential in the postsynaptic neurone.

In neuroscience, an excitatory postsynaptic potential (EPSP) is a postsynaptic potential that makes the postsynaptic neuron more likely to fire an action potential. This temporary depolarization of postsynaptic membrane potential, caused by the flow of positively charged ions into the postsynaptic cell, is a result of opening ligand-gated ion channels. These are the opposite of inhibitory postsynaptic potentials (IPSPs), which usually result from the flow of negative ions into the cell or positive ions out of the cell. EPSPs can also result from a decrease in outgoing positive charges, while IPSPs are sometimes caused by an increase in positive charge outflow. The flow of ions that causes an EPSP is an excitatory postsynaptic current (EPSC).
In neuroscience, a silent synapse is an excitatory glutamatergic synapse whose postsynaptic membrane contains NMDA-type glutamate receptors but no AMPA-type glutamate receptors. These synapses are named "silent" because normal AMPA receptor-mediated signaling is not present, rendering the synapse inactive under typical conditions. Silent synapses are typically considered to be immature glutamatergic synapses. As the brain matures, the relative number of silent synapses decreases. However, recent research on hippocampal silent synapses shows that while they may indeed be a developmental landmark in the formation of a synapse, that synapses can be "silenced" by activity, even once they have acquired AMPA receptors. Thus, silence may be a state that synapses can visit many times during their lifetimes.

An excitatory synapse is a synapse in which an action potential in a presynaptic neuron increases the probability of an action potential occurring in a postsynaptic cell. Neurons form networks through which nerve impulses travel, each neuron often making numerous connections with other cells. These electrical signals may be excitatory or inhibitory, and, if the total of excitatory influences exceeds that of the inhibitory influences, the neuron will generate a new action potential at its axon hillock, thus transmitting the information to yet another cell.

In excitotoxicity, nerve cells suffer damage or death when the levels of otherwise necessary and safe neurotransmitters such as glutamate become pathologically high, resulting in excessive stimulation of receptors. For example, when glutamate receptors such as the NMDA receptor or AMPA receptor encounter excessive levels of the excitatory neurotransmitter, glutamate, significant neuronal damage might ensue. Excess glutamate allows high levels of calcium ions (Ca2+) to enter the cell. Ca2+ influx into cells activates a number of enzymes, including phospholipases, endonucleases, and proteases such as calpain. These enzymes go on to damage cell structures such as components of the cytoskeleton, membrane, and DNA. In evolved, complex adaptive systems such as biological life it must be understood that mechanisms are rarely, if ever, simplistically direct. For example, NMDA in subtoxic amounts induces neuronal survival of otherwise toxic levels of glutamate.
Synaptogenesis is the formation of synapses between neurons in the nervous system. Although it occurs throughout a healthy person's lifespan, an explosion of synapse formation occurs during early brain development, known as exuberant synaptogenesis. Synaptogenesis is particularly important during an individual's critical period, during which there is a certain degree of synaptic pruning due to competition for neural growth factors by neurons and synapses. Processes that are not used, or inhibited during their critical period will fail to develop normally later on in life.

Ligand-gated ion channels (LICs, LGIC), also commonly referred to as ionotropic receptors, are a group of transmembrane ion-channel proteins which open to allow ions such as Na+, K+, Ca2+, and/or Cl− to pass through the membrane in response to the binding of a chemical messenger (i.e. a ligand), such as a neurotransmitter.
Molecular neuroscience is a branch of neuroscience that observes concepts in molecular biology applied to the nervous systems of animals. The scope of this subject covers topics such as molecular neuroanatomy, mechanisms of molecular signaling in the nervous system, the effects of genetics and epigenetics on neuronal development, and the molecular basis for neuroplasticity and neurodegenerative diseases. As with molecular biology, molecular neuroscience is a relatively new field that is considerably dynamic.

Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells. Glutamate is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
The ischemic (ischaemic) cascade is a series of biochemical reactions that are initiated in the brain and other aerobic tissues after seconds to minutes of ischemia. This is typically secondary to stroke, injury, or cardiac arrest due to heart attack. Most ischemic neurons that die do so due to the activation of chemicals produced during and after ischemia. The ischemic cascade usually goes on for two to three hours but can last for days, even after normal blood flow returns.
Gliotransmitters are chemicals released from glial cells that facilitate neuronal communication between neurons and other glial cells. They are usually induced from Ca2+ signaling, although recent research has questioned the role of Ca2+ in gliotransmitters and may require a revision of the relevance of gliotransmitters in neuronal signalling in general.

Potassium-chloride transporter member 5 is a neuron-specific chloride potassium symporter responsible for establishing the chloride ion gradient in neurons through the maintenance of low intracellular chloride concentrations. It is a critical mediator of synaptic inhibition, cellular protection against excitotoxicity and may also act as a modulator of neuroplasticity. Potassium-chloride transporter member 5 is also known by the names: KCC2 for its ionic substrates, and SLC12A5 for its genetic origin from the SLC12A5 gene in humans.
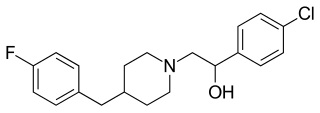
Eliprodil is an NMDA antagonist drug candidate which selectively inhibits the NR2B (GLUN2B) subtype NMDA receptor at submicromolar concentrations. Eliprodil failed a Phase III clinical trial for the treatment of acute ischemic stroke in 1996, sponsored by Synthélabo Recherche.

In neurophysiology, a dendritic spike refers to an action potential generated in the dendrite of a neuron. Dendrites are branched extensions of a neuron. They receive electrical signals emitted from projecting neurons and transfer these signals to the cell body, or soma. Dendritic signaling has traditionally been viewed as a passive mode of electrical signaling. Unlike its axon counterpart which can generate signals through action potentials, dendrites were believed to only have the ability to propagate electrical signals by physical means: changes in conductance, length, cross sectional area, etc. However, the existence of dendritic spikes was proposed and demonstrated by W. Alden Spencer, Eric Kandel, Rodolfo Llinás and coworkers in the 1960s and a large body of evidence now makes it clear that dendrites are active neuronal structures. Dendrites contain voltage-gated ion channels giving them the ability to generate action potentials. Dendritic spikes have been recorded in numerous types of neurons in the brain and are thought to have great implications in neuronal communication, memory, and learning. They are one of the major factors in long-term potentiation.

Nonsynaptic plasticity is a form of neuroplasticity that involves modification of ion channel function in the axon, dendrites, and cell body that results in specific changes in the integration of excitatory postsynaptic potentials and inhibitory postsynaptic potentials. Nonsynaptic plasticity is a modification of the intrinsic excitability of the neuron. It interacts with synaptic plasticity, but it is considered a separate entity from synaptic plasticity. Intrinsic modification of the electrical properties of neurons plays a role in many aspects of plasticity from homeostatic plasticity to learning and memory itself. Nonsynaptic plasticity affects synaptic integration, subthreshold propagation, spike generation, and other fundamental mechanisms of neurons at the cellular level. These individual neuronal alterations can result in changes in higher brain function, especially learning and memory. However, as an emerging field in neuroscience, much of the knowledge about nonsynaptic plasticity is uncertain and still requires further investigation to better define its role in brain function and behavior.

In neuroscience, glutamate refers to the anion of glutamic acid in its role as a neurotransmitter: a chemical that nerve cells use to send signals to other cells. It is by a wide margin the most abundant excitatory neurotransmitter in the vertebrate nervous system. It is used by every major excitatory function in the vertebrate brain, accounting in total for well over 90% of the synaptic connections in the human brain. It also serves as the primary neurotransmitter for some localized brain regions, such as cerebellum granule cells.