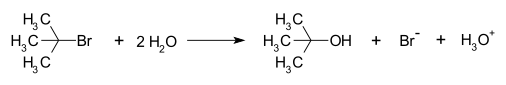
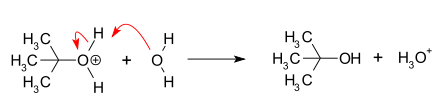
In chemistry, a nucleophile is a chemical species that forms bonds by donating an electron pair. All molecules and ions with a free pair of electrons or at least one pi bond can act as nucleophiles. Because nucleophiles donate electrons, they are Lewis bases.
A nucleophilic substitution is a class of chemical reactions in which an electron-rich chemical species replaces a functional group within another electron-deficient molecule. The molecule that contains the electrophile and the leaving functional group is called the substrate.

An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.
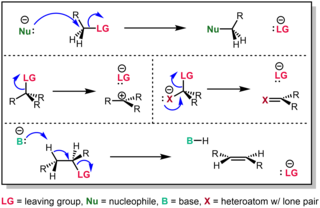
In chemistry, a leaving group is defined by the IUPAC as an atom or group of atoms that detaches from the main or residual part of a substrate during a reaction or elementary step of a reaction. However, in common usage, the term is often limited to a fragment that departs with a pair of electrons in heterolytic bond cleavage. In this usage, a leaving group is a less formal but more commonly used synonym of the term nucleofuge. In this context, leaving groups are generally anions or neutral species, departing from a neutral or cationic substrates, respectively, though in rare cases, cations leaving from a dicationic substrate are also known. A species' ability to serve as a leaving group depends on its ability to stabilize the additional electron density that results from bond heterolysis. Common anionic leaving groups are halides such as Cl−, Br−, and I−, and sulfonate esters such as tosylate (TsO−), while water (H2O), alcohols (HOR), and amines (R3N) are common neutral leaving groups.
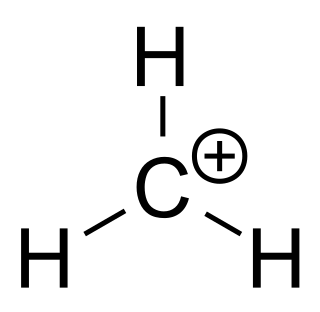
A carbocation is an ion with a positively charged carbon atom. Among the simplest examples are the methenium CH+
3, methanium CH+
5 and vinyl C
2H+
3 cations. Occasionally, carbocations that bear more than one positively charged carbon atom are also encountered.
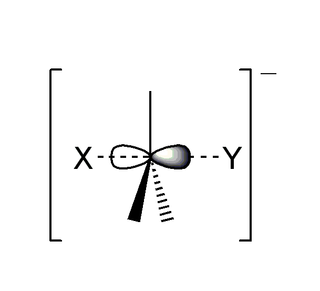
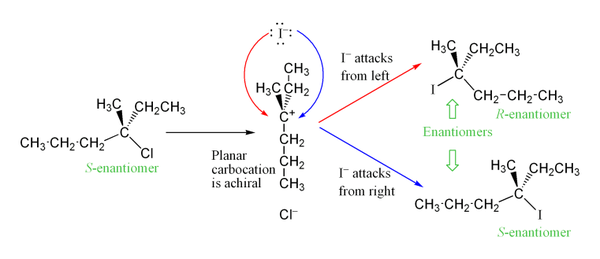
The SN2 reaction is a type of reaction mechanism that is common in organic chemistry. In this mechanism, one bond is broken and one bond is formed synchronously, i.e., in one step. SN2 is a kind of nucleophilic substitution reaction mechanism, the name referring to the Hughes-Ingold symbol of the mechanism. Since two reacting species are involved in the slow (rate-determining) step, this leads to the term substitution nucleophilic (bi-molecular) or SN2; the other major kind is SN1. Many other more specialized mechanisms describe substitution reactions.
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.
A substitution reaction is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group. Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
In chemical kinetics, the overall rate of a reaction is often approximately determined by the slowest step, known as the rate-determining step or rate-limiting step. For a given reaction mechanism, the prediction of the corresponding rate equation is often simplified by using this approximation of the rate-determining step.

The Appel reaction is an organic reaction that converts an alcohol into an alkyl chloride using triphenylphosphine and carbon tetrachloride. The use of carbon tetrabromide or bromine as a halide source will yield alkyl bromides, whereas using carbon tetraiodide, methyl iodide or iodine gives alkyl iodides. The reaction is credited to and named after Rolf Appel, it had however been described earlier. The use of this reaction is becoming less common, due to carbon tetrachloride being restricted under the Montreal protocol.
Solvolysis is a type of nucleophilic substitution (SN1/SN2) or elimination where the nucleophile is a solvent molecule. Characteristic of SN1 reactions, solvolysis of a chiral reactant affords the racemate. Sometimes however, the stereochemical course is complicated by intimate ion pairs, whereby the leaving anion remains close to the carbocation, effectively shielding it from an attack by the nucleophile. Particularly fast reactions can occur by neighbour group participation, with nonclassical ions as intermediates or transition states.
In chemistry the intimate ion pair concept introduced by Saul Winstein describes the interactions between a cation, anion and surrounding solvent molecules. In ordinary aqueous solutions of inorganic salts an ion is completely solvated and shielded from the counterion. In less polar solvents two ions can still be connected to some extent. In a tight or intimate or contact ion pair there are no solvent molecules between the two ions. When solvation increases, ionic bonding decreases and a loose or solvent-shared ion pair results. The ion pair concept explains stereochemistry in solvolysis.
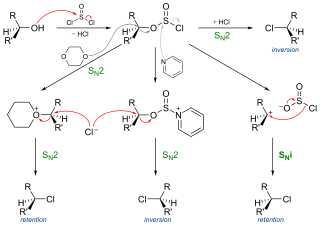
SNi or Substitution Nucleophilic intramolecular stands for a specific but not often encountered nucleophilic aliphatic substitution reaction mechanism. The name was introduced by Cowdrey et al. in 1937 to label nucleophilic reactions which occur with retention of configuration, but later was employed to describe various reactions that proceed with similar mechanism.
In organic chemistry, neighbouring group participation has been defined by the International Union of Pure and Applied Chemistry (IUPAC) as the interaction of a reaction centre with a lone pair of electrons in an atom or the electrons present in a pi bond contained within the parent molecule but not conjugated with the reaction centre. When NGP is in operation it is normal for the reaction rate to be increased. It is also possible for the stereochemistry of the reaction to be abnormal when compared with a normal reaction. While it is possible for neighbouring groups to influence many reactions in organic chemistry this page is limited to neighbouring group effects seen with carbocations and SN2 reactions.

A Grignard reagent or Grignard compound is a chemical compound with the generic formula R−Mg−X, where X is a halogen and R is an organic group, normally an alkyl or aryl. Two typical examples are methylmagnesium chloride Cl−Mg−CH3 and phenylmagnesium bromide (C6H5)−Mg−Br. They are a subclass of the organomagnesium compounds.
In physical organic chemistry, the Grunwald–Winstein equation is a linear free energy relationship between relative rate constants and the ionizing power of various solvent systems, describing the effect of solvent as nucleophile on different substrates. The equation, which was developed by Ernest Grunwald and Saul Winstein in 1948, could be written
In chemistry, solvent effects are the influence of a solvent on chemical reactivity or molecular associations. Solvents can have an effect on solubility, stability and reaction rates and choosing the appropriate solvent allows for thermodynamic and kinetic control over a chemical reaction.
Living cationic polymerization is a living polymerization technique involving cationic propagating species. It enables the synthesis of very well defined polymers and of polymers with unusual architecture such as star polymers and block copolymers and living cationic polymerization is therefore as such of commercial and academic interest.
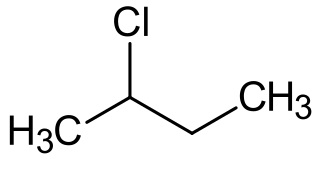
2-Chlorobutane is a compound with formula C4H9Cl. It is also called sec-butyl chloride. It is a colorless, volatile liquid at room temperature that is not miscible in water.
Ether cleavage refers to chemical substitution reactions that lead to the cleavage of ethers. Due to the high chemical stability of ethers, the cleavage of the C-O bond is uncommon in the absence of specialized reagents or under extreme conditions.