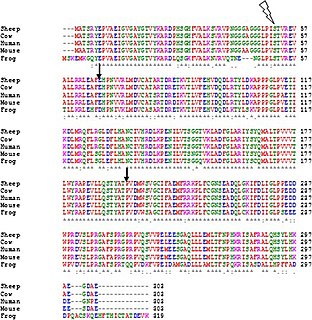
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the edit distance cost between strings in a natural language or in financial data.
In bioinformatics and evolutionary biology, a substitution matrix describes the rate at which one character in a sequence changes to other character states over time. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where the similarity between sequences depends on their divergence time and the substitution rates as represented in the matrix.
In bioinformatics, BLAST is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold.
In computational linguistics and computer science, edit distance is a way of quantifying how dissimilar two strings are to one another by counting the minimum number of operations required to transform one string into the other. Edit distances find applications in natural language processing, where automatic spelling correction can determine candidate corrections for a misspelled word by selecting words from a dictionary that have a low distance to the word in question. In bioinformatics, it can be used to quantify the similarity of DNA sequences, which can be viewed as strings of the letters A, C, G and T.
In statistics and related fields, a similarity measure or similarity function is a real-valued function that quantifies the similarity between two objects. Although no single definition of a similarity measure exists, usually such measures are in some sense the inverse of distance metrics: they take on large values for similar objects and either zero or a negative value for very dissimilar objects. For example, if two pieces of data have close x, y coordinates, then their “similarity” score, the likelihood that they are similar, will be much higher than two data points with more space between them. In the context of cluster analysis, Frey and Dueck suggest defining a similarity measure

The Euclidean minimum spanning tree or EMST is a minimum spanning tree of a set of n points in the plane, where the weight of the edge between each pair of points is the Euclidean distance between those two points. In simpler terms, an EMST connects a set of dots using lines such that the total length of all the lines is minimized and any dot can be reached from any other by following the lines.
Clustal is a series of widely used computer programs used in Bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm are also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
In information theory and computer science, the Damerau–Levenshtein distance is a string metric for measuring the edit distance between two sequences. Informally, the Damerau–Levenshtein distance between two words is the minimum number of operations required to change one word into the other.

In computer science, approximate string matching is the technique of finding strings that match a pattern approximately. The problem of approximate string matching is typically divided into two sub-problems: finding approximate substring matches inside a given string and finding dictionary strings that match the pattern approximately.
Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. For example, these techniques have been used to explore the family tree of hominid species and the relationships between specific genes shared by many types of organisms. Traditional phylogenetics relies on morphological data obtained by measuring and quantifying the phenotypic properties of representative organisms, while the more recent field of molecular phylogenetics uses nucleotide sequences encoding genes or amino acid sequences encoding proteins as the basis for classification. Many forms of molecular phylogenetics are closely related to and make extensive use of sequence alignment in constructing and refining phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The phylogenetic trees constructed by computational methods are unlikely to perfectly reproduce the evolutionary tree that represents the historical relationships between the species being analyzed. The historical species tree may also differ from the historical tree of an individual homologous gene shared by those species.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.
Bayesian inference of phylogeny uses a likelihood function to create a quantity called the posterior probability of trees using a model of evolution, based on some prior probabilities, producing the most likely phylogenetic tree for the given data. The Bayesian approach has become popular due to advances in computing speeds and the integration of Markov chain Monte Carlo (MCMC) algorithms. Bayesian inference has a number of applications in molecular phylogenetics and systematics.
Least squares inference in phylogeny generates a phylogenetic tree based on an observed matrix of pairwise genetic distances and optionally a weight matrix. The goal is to find a tree which satisfies the distance constraints as best as possible.

UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.
The Robinson–Foulds metric is a way to measure the distance between unrooted phylogenetic trees. It is defined as where A is the number of partitions of data implied by the first tree but not the second tree and B is the number of partitions of data implied by the second tree but not the first tree. The partitions are calculated for each tree by removing each branch. Thus, the number of eligible partitions for each tree is equal to the number of branches in that tree. The Robinson–Foulds metric is also known as the symmetric difference metric.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.
Horizontal or lateral gene transfer is the transmission of portions of genomic DNA between organisms through a process decoupled from vertical inheritance. In the presence of HGT events, different fragments of the genome are the result of different evolutionary histories. This can therefore complicate the investigations of evolutionary relatedness of lineages and species. Also, as HGT can bring into genomes radically different genotypes from distant lineages, or even new genes bearing new functions, it is a major source of phenotypic innovation and a mechanism of niche adaptation. For example, of particular relevance to human health is the lateral transfer of antibiotic resistance and pathogenicity determinants, leading to the emergence of pathogenic lineages.