
Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.

Sir John Anthony Pople was a British theoretical chemist who was awarded the Nobel Prize in Chemistry with Walter Kohn in 1998 for his development of computational methods in quantum chemistry.
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.

Robert Ghormley Parr was an American theoretical chemist who was a professor of chemistry at the University of North Carolina at Chapel Hill.

Theoretical chemistry is the branch of chemistry which develops theoretical generalizations that are part of the theoretical arsenal of modern chemistry: for example, the concepts of chemical bonding, chemical reaction, valence, the surface of potential energy, molecular orbitals, orbital interactions, and molecule activation.
A linear combination of atomic orbitals or LCAO is a quantum superposition of atomic orbitals and a technique for calculating molecular orbitals in quantum chemistry. In quantum mechanics, electron configurations of atoms are described as wavefunctions. In a mathematical sense, these wave functions are the basis set of functions, the basis functions, which describe the electrons of a given atom. In chemical reactions, orbital wavefunctions are modified, i.e. the electron cloud shape is changed, according to the type of atoms participating in the chemical bond.
Gaussian is a general purpose computational chemistry software package initially released in 1970 by John Pople and his research group at Carnegie Mellon University as Gaussian 70. It has been continuously updated since then. The name originates from Pople's use of Gaussian orbitals to speed up molecular electronic structure calculations as opposed to using Slater-type orbitals, a choice made to improve performance on the limited computing capacities of then-current computer hardware for Hartree–Fock calculations. The current version of the program is Gaussian 16. Originally available through the Quantum Chemistry Program Exchange, it was later licensed out of Carnegie Mellon University, and since 1987 has been developed and licensed by Gaussian, Inc.
Q-Chem is a general-purpose electronic structure package featuring a variety of established and new methods implemented using innovative algorithms that enable fast calculations of large systems on various computer architectures, from laptops and regular lab workstations to midsize clusters, HPCC, and cloud computing using density functional and wave-function based approaches. It offers an integrated graphical interface and input generator; a large selection of functionals and correlation methods, including methods for electronically excited states and open-shell systems; solvation models; and wave-function analysis tools. In addition to serving the computational chemistry community, Q-Chem also provides a versatile code development platform.
Psi is an ab initio computational chemistry package originally written by the research group of Henry F. Schaefer, III. Utilizing Psi, one can perform a calculation on a molecular system with various kinds of methods such as Hartree-Fock, Post-Hartree–Fock electron correlation methods, and density functional theory. The program can compute energies, optimize molecular geometries, and compute vibrational frequencies. The major part of the program is written in C++, while Python API is also available, which allows users to perform complex computations or automate tasks easily.
In molecular physics, the Pariser–Parr–Pople method applies semi-empirical quantum mechanical methods to the quantitative prediction of electronic structures and spectra, in molecules of interest in the field of organic chemistry. Previous methods existed—such as the Hückel method which led to Hückel's rule—but were limited in their scope, application and complexity, as is the Extended Hückel method.
In computational chemistry, post–Hartree–Fock (post-HF) methods are the set of methods developed to improve on the Hartree–Fock (HF), or self-consistent field (SCF) method. They add electron correlation which is a more accurate way of including the repulsions between electrons than in the Hartree–Fock method where repulsions are only averaged.
General Atomic and Molecular Electronic Structure System (GAMESS-UK) is a computer software program for computational chemistry. The original code split in 1981 into GAMESS-UK and GAMESS (US) variants, which now differ significantly. Many of the early developments in the UK version arose from the earlier UK based ATMOL program, which, unlike GAMESS, lacked analytical gradients for geometry optimisation.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Semi-empirical quantum chemistry methods are based on the Hartree–Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree–Fock method without the approximations is too expensive. The use of empirical parameters appears to allow some inclusion of electron correlation effects into the methods.
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene. The background is described by Parr. Ab initio means "from first principles" or "from the beginning", implying that the only inputs into an ab initio calculation are physical constants. Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to solve a range of problems and their importance is highlighted by the awarding of the Nobel prize to John Pople and Walter Kohn.
Axel Dieter Becke is a physical chemist and Professor of Chemistry at Dalhousie University, Canada. He is a leading researcher in the application of density functional theory (DFT) to molecules.
CP2K is a freely available (GPL) quantum chemistry and solid state physics program package, written in Fortran 2008, to perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. It provides a general framework for different methods: density functional theory (DFT) using a mixed Gaussian and plane waves approach (GPW) via LDA, GGA, MP2, or RPA levels of theory, classical pair and many-body potentials, semi-empirical and tight-binding Hamiltonians, as well as Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid schemes relying on the Gaussian Expansion of the Electrostatic Potential (GEEP). The Gaussian and Augmented Plane Waves method (GAPW) as an extension of the GPW method allows for all-electron calculations. CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method.
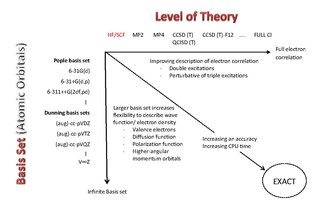
A Pople diagram or Pople's Diagram is a diagram which describes the relationship between various calculation methods in computational chemistry. It was initially introduced in January 1965 by Sir John Pople,, during the Symposium of Atomic and Molecular Quantum Theory in Florida. The Pople Diagram can be either 2-dimensional or 3-dimensional, with the axes representing ab initio methods, basis sets and treatment of relativity. The diagram attempts to balance calculations by giving all aspects of a computation equal weight.
Kim K. Baldridge is an American theoretical and computational chemist who works to develop quantum mechanical methodologies and apply quantum chemical methods to problems in life sciences, materials science, and general studies. She is professor and vice dean in the School of Pharmaceutical Science and Technology of Tianjin University in China, where she also directs the High Performance Computing Center.