Molecular dynamics (MD) is a computer simulation method for studying the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic evolution of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanics force fields. The method was originally developed within the field of theoretical physics in the late 1950s but is applied today mostly in chemical physics, materials science and the modelling of biomolecules.
In computational physics and chemistry, the Hartree–Fock (HF) method is a method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
In solid-state physics, the electronic band structure of a solid describes the range of energies an electron within the solid may have and ranges of energy that it may not have.

SIESTA is an original method and its computer program implementation, to perform efficient electronic structure calculations and ab initio molecular dynamics simulations of molecules and solids. SIESTA's efficiency stems from the use of strictly localized basis sets and from the implementation of linear-scaling algorithms which can be applied to suitable systems. A very important feature of the code is that its accuracy and cost can be tuned in a wide range, from quick exploratory calculations to highly accurate simulations matching the quality of other approaches, such as plane-wave and all-electron methods.
octopus is a software package for performing Kohn–Sham density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations.
X-ray absorption near edge structure (XANES), also known as near edge X-ray absorption fine structure (NEXAFS), is a type of absorption spectroscopy that indicates the features in the X-ray absorption spectra (XAS) of condensed matter due to the photoabsorption cross section for electronic transitions from an atomic core level to final states in the energy region of 50–100 eV above the selected atomic core level ionization energy, where the wavelength of the photoelectron is larger than the interatomic distance between the absorbing atom and its first neighbour atoms.
The fragment molecular orbital method (FMO) is a computational method that can compute very large molecular systems with thousands of atoms using ab initio quantum-chemical wave functions.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.
The Reverse Monte Carlo (RMC) modelling method is a variation of the standard Metropolis-Hastings algorithm to solve an inverse problem whereby a model is adjusted until its parameters have the greatest consistency with experimental data. Inverse problems are found in many branches of science and mathematics, but this approach is probably best known for its applications in condensed matter physics and solid state chemistry.
In computational physics, variational Monte Carlo (VMC) is a quantum Monte Carlo method that applies the variational method to approximate the ground state of a quantum system.
In the field of computational chemistry, energy minimization is the process of finding an arrangement in space of a collection of atoms where, according to some computational model of chemical bonding, the net inter-atomic force on each atom is acceptably close to zero and the position on the potential energy surface (PES) is a stationary point. The collection of atoms might be a single molecule, an ion, a condensed phase, a transition state or even a collection of any of these. The computational model of chemical bonding might, for example, be quantum mechanics.
PARSEC is a package designed to perform electronic structure calculations of solids and molecules using density functional theory (DFT). The acronym stands for Pseudopotential Algorithm for Real-Space Electronic Calculations. It solves the Kohn–Sham equations in real space, without the use of explicit basis sets.
Yambo is a computer software package for studying many-body theory aspects of solids and molecule systems. It calculates the excited state properties of physical systems from first principles, e.g., from quantum mechanics law without the use of empirical data. Parts of it are open-source software released under a GNU General Public License (GPL).
Quantum ESPRESSO is a software suite for ab initio quantum chemistry methods of electronic-structure calculation and materials modeling, distributed for free under the GNU General Public License. It is based on Density Functional Theory, plane wave basis sets, and pseudopotentials. ESPRESSO is an acronym for opEn-Source Package for Research in Electronic Structure, Simulation, and Optimization.
BigDFT is a free software package for physicists and chemists, distributed under the GNU General Public License, whose main program allows the total energy, charge density, and electronic structure of systems made of electrons and nuclei to be calculated within density functional theory (DFT), using pseudopotentials, and a wavelet basis.
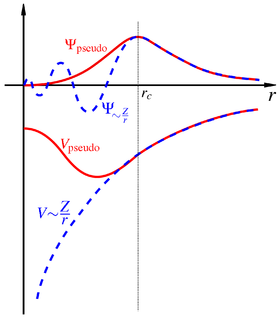
The projector augmented wave method (PAW) is a technique used in ab initio electronic structure calculations. It is a generalization of the pseudopotential and linear augmented-plane-wave methods, and allows for density functional theory calculations to be performed with greater computational efficiency.
The prediction of crystal properties by numerical simulation has become commonplace in the last 20 years as computers have grown more powerful and theoretical techniques more sophisticated. High accuracy prediction of elastic, electronic, transport and phase properties is possible with modern methods.

FHI-aims is a shared-source software package for computational molecular and materials science written in Fortran. It uses density functional theory and many-body perturbation theory to simulate chemical and physical properties of atoms, molecules, nanostructures, soldis, and surfaces. Originally developed at the Fritz Haber Institute in Berlin the ongoing development of the FHI-aims source code is now driven by a world-wide community of collaborating research institutions.