External links
| | This bioinformatics-related article is a stub. You can help Wikipedia by expanding it. |
Align-m is a multiple sequence alignment program written by Ivo Van Walle.
Align-m has the ability to accomplish the following tasks:
| | This bioinformatics-related article is a stub. You can help Wikipedia by expanding it. |

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
In bioinformatics and evolutionary biology, a substitution matrix describes the frequency at which a character in a nucleotide sequence or a protein sequence changes to other character states over evolutionary time. The information is often in the form of log odds of finding two specific character states aligned and depends on the assumed number of evolutionary changes or sequence dissimilarity between compared sequences. It is an application of a stochastic matrix. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where they are used to calculate similarity scores between the aligned sequences.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.

The Needleman–Wunsch algorithm is an algorithm used in bioinformatics to align protein or nucleotide sequences. It was one of the first applications of dynamic programming to compare biological sequences. The algorithm was developed by Saul B. Needleman and Christian D. Wunsch and published in 1970. The algorithm essentially divides a large problem into a series of smaller problems, and it uses the solutions to the smaller problems to find an optimal solution to the larger problem. It is also sometimes referred to as the optimal matching algorithm and the global alignment technique. The Needleman–Wunsch algorithm is still widely used for optimal global alignment, particularly when the quality of the global alignment is of the utmost importance. The algorithm assigns a score to every possible alignment, and the purpose of the algorithm is to find all possible alignments having the highest score.
A Gap penalty is a method of scoring alignments of two or more sequences. When aligning sequences, introducing gaps in the sequences can allow an alignment algorithm to match more terms than a gap-less alignment can. However, minimizing gaps in an alignment is important to create a useful alignment. Too many gaps can cause an alignment to become meaningless. Gap penalties are used to adjust alignment scores based on the number and length of gaps. The five main types of gap penalties are constant, linear, affine, convex, and profile-based.
Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm is also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
In molecular biology, protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.

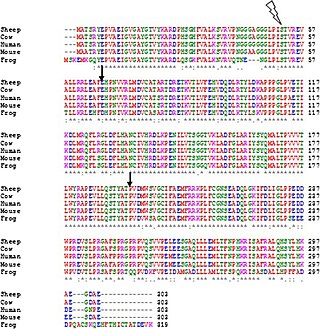
Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
T-Coffee is a multiple sequence alignment software using a progressive approach. It generates a library of pairwise alignments to guide the multiple sequence alignment. It can also combine multiple sequences alignments obtained previously and in the latest versions can use structural information from PDB files (3D-Coffee). It has advanced features to evaluate the quality of the alignments and some capacity for identifying occurrence of motifs (Mocca). It produces alignment in the aln format (Clustal) by default, but can also produce PIR, MSF, and FASTA format. The most common input formats are supported.
ProbCons is an open source probabilistic consistency-based multiple alignment of amino acid sequences. It is one of the most efficient protein multiple sequence alignment programs, since it has repeatedly demonstrated a statistically significant advantage in accuracy over similar tools, including Clustal and MAFFT.
MUltiple Sequence Comparison by Log-Expectation (MUSCLE) is computer software for multiple sequence alignment of protein and nucleotide sequences. It is licensed as public domain. The method was published by Robert C. Edgar in two papers in 2004. The first paper, published in Nucleic Acids Research, introduced the sequence alignment algorithm. The second paper, published in BMC Bioinformatics, presented more technical details.
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements. It is an annotated, open access database originally developed at the Wellcome Trust Sanger Institute in collaboration with Janelia Farm, and currently hosted at the European Bioinformatics Institute. Rfam is designed to be similar to the Pfam database for annotating protein families.
ESyPred3D is an automated homology modeling program. Alignments are obtained by combining, weighting and screening the results of several multiple alignment programs. The final three-dimensional structure is built using the modeling package MODELLER.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and macOS.
VISTA is a collection of databases, tools, and servers that permit extensive comparative genomics analyses.
FSA is a multiple sequence alignment program for aligning many proteins or RNAs or long genomic DNA sequences. Along with MUSCLE and MAFFT, FSA is one of the few sequence alignment programs which can align datasets of hundreds or thousands of sequences. FSA uses a different optimization criterion which allows it to more reliably identify non-homologous sequences than these other programs, although this increased accuracy comes at the cost of decreased speed.
Phyloscan is a web service for DNA sequence analysis that is free and open to all users. For locating matches to a user-specified sequence motif for a regulatory binding site, Phyloscan provides a statistically sensitive scan of user-supplied mixed aligned and unaligned DNA sequence data. Phyloscan's strength is that it brings together
Phylo is an experimental video game about multiple sequence alignment optimisation. Developed by the McGill Centre for Bioinformatics, it was originally released as a free Flash game in November 2010. Designed as a game with a purpose, players solve pattern-matching puzzles that represent nucleotide sequences of different phylogenetic taxa to optimize alignments over a computer algorithm. By aligning together each nucleotide sequence, represented as differently coloured blocks, players attempt to create the highest point value score for each set of sequences by matching as many colours as possible and minimizing gaps.
SAMtools is a set of utilities for interacting with and post-processing short DNA sequence read alignments in the SAM, BAM and CRAM formats, written by Heng Li. These files are generated as output by short read aligners like BWA. Both simple and advanced tools are provided, supporting complex tasks like variant calling and alignment viewing as well as sorting, indexing, data extraction and format conversion. SAM files can be very large, so compression is used to save space. SAM files are human-readable text files, and BAM files are simply their binary equivalent, whilst CRAM files are a restructured column-oriented binary container format. BAM files are typically compressed and more efficient for software to work with than SAM. SAMtools makes it possible to work directly with a compressed BAM file, without having to uncompress the whole file. Additionally, since the format for a SAM/BAM file is somewhat complex - containing reads, references, alignments, quality information, and user-specified annotations - SAMtools reduces the effort needed to use SAM/BAM files by hiding low-level details.