The bibliome is the totality of biological text corpus. This term was coined around 2000 in EBI (European Bioinformatics Institute) to denote the importance of biological text information. Similar terms that have been less frequently used are literaturome and textome.[citation needed] By approximate analogy to widely used terms like genome, metabolome, proteome, and transcriptome, this -ome would properly refer to the literature of a specified or contextually implied field, hence: biological bibliome, political bibliome, etc.[citation needed] However the term has not (yet) been applied outside the biological and medical sciences so it currently by default applies just to the biomedical fields. It would make little sense to apply it to a particular body of texts such as MEDLINE, despite a natural analogy that might seem to suggest this: the terms genome, proteome, channelome, metabolome, and transcriptome all usually assume a specific organism or cell set and (except for genome) a specific time point. The reason following this analogy would make little sense is that there is already an established term for this purpose, corpus.
In linguistics, a corpus or text corpus is a large and structured set of texts. In corpus linguistics, they are used to do statistical analysis and hypothesis testing, checking occurrences or validating linguistic rules within a specific language territory.
The European Bioinformatics Institute (EMBL-EBI) is an IGO which as part of the European Molecular Biology Laboratory (EMBL) family focuses on research and services in bioinformatics.
A biologist is a scientist who has specialized knowledge in the field of biology, the scientific study of life. Biologists involved in fundamental research attempt to explore and further explain the underlying mechanisms that govern the functioning of living matter. Biologists involved in applied research attempt to develop or improve more specific processes and understanding, in fields such as medicine and industry.
A computer scientist is a person who has acquired the knowledge of computer science, the study of the theoretical foundations of information and computation and their application.
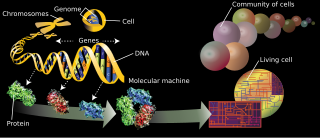
In biology, a gene is a sequence of nucleotides in DNA or RNA that codes for a molecule that has a function. During gene expression, the DNA is first copied into RNA. The RNA can be directly functional or be the intermediate template for a protein that performs a function. The transmission of genes to an organism's offspring is the basis of the inheritance of phenotypic trait. These genes make up different DNA sequences called genotypes. Genotypes along with environmental and developmental factors determine what the phenotypes will be. Most biological traits are under the influence of polygenes as well as gene–environment interactions. Some genetic traits are instantly visible, such as eye color or number of limbs, and some are not, such as blood type, risk for specific diseases, or the thousands of basic biochemical processes that constitute life.
Online applications
EAGLi is a biomedical retrieval engine for MEDLINE. GOCat is an Automatic GO categorizer/browser to help functional annotation out of biomedical texts; also useful to functionally characterize protein and gene names lists generated by high-throughput experiments.
In computing, a Digital Object Identifier or DOI is a persistent identifier or handle used to identify objects uniquely, standardized by the International Organization for Standardization (ISO). An implementation of the Handle System, DOIs are in wide use mainly to identify academic, professional, and government information, such as journal articles, research reports and data sets, and official publications though they also have been used to identify other types of information resources, such as commercial videos.
PubMed Central (PMC) is a free digital repository that archives publicly accessible full-text scholarly articles that have been published within the biomedical and life sciences journal literature. As one of the major research databases within the suite of resources that have been developed by the National Center for Biotechnology Information (NCBI), PubMed Central is much more than just a document repository. Submissions into PMC undergo an indexing and formatting procedure which results in enhanced metadata, medical ontology, and unique identifiers which all enrich the XML structured data for each article on deposit. Content within PMC can easily be interlinked to many other NCBI databases and accessed via Entrez search and retrieval systems, further enhancing the public's ability to freely discover, read and build upon this portfolio of biomedical knowledge.
This bioinformatics-related article is a stub. You can help Wikipedia by expanding it.
Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data. As an interdisciplinary field of science, bioinformatics combines biology, computer science, information engineering, mathematics and statistics to analyze and interpret biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.
The proteome is the entire set of proteins that is, or can be, expressed by a genome, cell, tissue, or organism at a certain time. It is the set of expressed proteins in a given type of cell or organism, at a given time, under defined conditions. Proteomics is the study of the proteome.
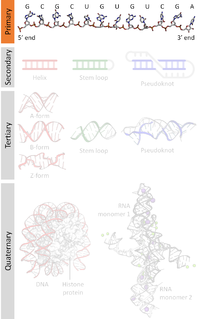
A nucleic acid sequence is a succession of letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
Biological databases are libraries of life sciences information, collected from scientific experiments, published literature, high-throughput experiment technology, and computational analysis. They contain information from research areas including genomics, proteomics, metabolomics, microarray gene expression, and phylogenetics. Information contained in biological databases includes gene function, structure, localization, clinical effects of mutations as well as similarities of biological sequences and structures.
The English-language neologism omics informally refers to a field of study in biology ending in -omics, such as genomics, proteomics or metabolomics. Omics aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism or organisms.
Metabolomics is the scientific study of chemical processes involving metabolites, the small molecule intermediates and products of metabolism. Specifically, metabolomics is the "systematic study of the unique chemical fingerprints that specific cellular processes leave behind", the study of their small-molecule metabolite profiles. The metabolome represents the complete set of metabolites in a biological cell, tissue, organ or organism, which are the end products of cellular processes. mRNA gene expression data and proteomic analyses reveal the set of gene products being produced in the cell, data that represents one aspect of cellular function. Conversely, metabolic profiling can give an instantaneous snapshot of the physiology of that cell, and thus, metabolomics provides a direct "functional readout of the physiological state" of an organism. One of the challenges of systems biology and functional genomics is to integrate genomics, transcriptomic, proteomic, and metabolomic information to provide a better understanding of cellular biology.
The transcriptome is the set of all RNA molecules in one cell or a population of cells. It is sometimes used to refer to all RNAs, or just mRNA, depending on the particular experiment. It differs from the exome in that it includes only those RNA molecules found in a specified cell population, and usually includes the amount or concentration of each RNA molecule in addition to the molecular identities.
The metabolome refers to the complete set of small-molecule chemicals found within a biological sample. The biological sample can be a cell, a cellular organelle, an organ, a tissue, a tissue extract, a biofluid or an entire organism. The small molecule chemicals found in a given metabolome may include both endogenous metabolites that are naturally produced by an organism as well as exogenous chemicals that are not naturally produced by an organism.
Gene ontology (GO) is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species. More specifically, the project aims to: 1) maintain and develop its controlled vocabulary of gene and gene product attributes; 2) annotate genes and gene products, and assimilate and disseminate annotation data; and 3) provide tools for easy access to all aspects of the data provided by the project, and to enable functional interpretation of experimental data using the GO, for example via enrichment analysis. GO is part of a larger classification effort, the Open Biomedical Ontologies (OBO).
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.
Biomedical text mining refers to the methods and study of how text mining may be applied to texts and literature of the biomedical and molecular biology domains. As a field of research, biomedical text mining incorporates ideas from natural language processing, bioinformatics, medical informatics and computational linguistics. The strategies developed through studies in this field are frequently applied to the biomedical and molecular biology literature available through services such as PubMed.
Open Biomedical Ontologies is an effort to create controlled vocabularies for shared use across different biological and medical domains. As of 2006, OBO forms part of the resources of the U.S. National Center for Biomedical Ontology where it will form a central element of the NCBO's BioPortal.
Integrative bioinformatics is a discipline of bioinformatics that focuses on problems of data integration for the life sciences.
The National Centre for Text Mining (NaCTeM) is a publicly funded text mining (TM) centre. It was established to provide support, advice, and information on TM technologies and to disseminate information from the larger TM community, while also providing tailored services and tools in response to the requirements of the United Kingdom academic community.
Co-occurrence networks are generally used to provide a graphic visualization of potential relationships between people, organizations, concepts, biological organisms like bacteria or other entities represented within written material. The generation and visualization of co-occurrence networks has become practical with the advent of electronically stored text compliant to text mining.
Medical biology is a field of biology that has practical applications in medicine, health care and laboratory diagnostics. It includes many biomedical disciplines and areas of specialty that typically contain the "bio-" prefix such as:
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Translational bioinformatics (TBI) is an emerging field in the study of health informatics, focused on the convergence of molecular bioinformatics, biostatistics, statistical genetics and clinical informatics. Its focus is on applying informatics methodology to the increasing amount of biomedical and genomic data to formulate knowledge and medical tools, which can be utilized by scientists, clinicians, and patients. Furthermore, it involves applying biomedical research to improve human health through the use of computer-based information system. TBI employs data mining and analyzing biomedical informatics in order to generate clinical knowledge for application. Clinical knowledge includes finding similarities in patient populations, interpreting biological information to suggest therapy treatments and predict health outcomes.
In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text-mining derived associations including Mendelian, complex and environmental diseases.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.