
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Turricephaly is a type of cephalic disorder where the head appears tall with a small length and width. It is due to premature closure of the coronal suture plus any other suture, like the lambdoid, or it may be used to describe the premature fusion of all sutures. It should be differentiated from Crouzon syndrome. Oxycephaly is a form of turricephaly where the head is cone-shaped, and is the most severe of the craniosynostoses.
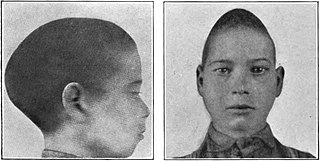
Scaphocephaly, or sagittal craniosynostosis, is a type of cephalic disorder which occurs when there is a premature fusion of the sagittal suture. Premature closure results in limited lateral expansion of the skull resulting in a characteristic long, narrow head. The skull base is typically spared.

Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Since the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects.

Apert syndrome is a form of acrocephalosyndactyly, a congenital disorder characterized by malformations of the skull, face, hands and feet. It is classified as a branchial arch syndrome, affecting the first branchial arch, the precursor of the maxilla and mandible. Disturbances in the development of the branchial arches in fetal development create lasting and widespread effects.

Craniosynostosis is a condition in which one or more of the fibrous sutures in a young infant's skull prematurely fuses by turning into bone (ossification), thereby changing the growth pattern of the skull. Because the skull cannot expand perpendicular to the fused suture, it compensates by growing more in the direction parallel to the closed sutures. Sometimes the resulting growth pattern provides the necessary space for the growing brain, but results in an abnormal head shape and abnormal facial features. In cases in which the compensation does not effectively provide enough space for the growing brain, craniosynostosis results in increased intracranial pressure leading possibly to visual impairment, sleeping impairment, eating difficulties, or an impairment of mental development combined with a significant reduction in IQ.

Carpenter syndrome, also called acrocephalopolysyndactyly type II, is an extremely rare autosomal recessive congenital disorder characterized by craniofacial malformations, obesity, syndactyly, and polydactyly. Acrocephalopolysyndactyly is a variation of acrocephalosyndactyly that presents with polydactyly.

Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.

Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux).

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.
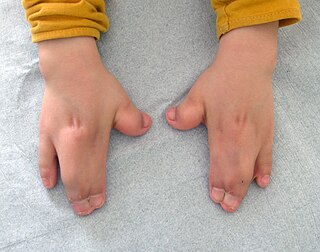
Acrocephalosyndactyly is a group of autosomal dominant congenital disorders characterized by craniofacial (craniosynostosis) and hand and foot (syndactyly) abnormalities. When polydactyly is present, the classification is acrocephalopolysyndactyly. Acrocephalosyndactyly is mainly diagnosed postnatally, although prenatal diagnosis is possible if the mutation is known to be within the family genome. Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) is a very rare genetic disorder. This disorder is one that affects bone growth and is characterized by skeletal, brain, and skin abnormalities. Those affected by the disorder are severely short in height and commonly possess shorter arms and legs. In addition, the bones of the legs are often bowed and the affected have smaller chests with shorter rib bones, along with curved collarbones. Other symptoms of the disorder include broad fingers and extra folds of skin on the arms and legs. Developmentally, many individuals who suffer from the disorder show a higher level in delays and disability. Seizures are also common due to structural abnormalities of the brain. Those affected may also suffer with apnea, the slowing or loss of breath for short periods of time.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.

Metacarpal synostosis is a rare congenital difference which is characterized by the fusion of 2 metacarpals of the hand, which are usually shortened. It is most commonly seen as a fusion of the 4th and 5th metacarpals. It is a type of non-syndromic syndactyly/synostosis. Autosomal dominant and X-linked recessive inheritance patterns have been reported.

Heart-hand syndrome, Slovenian type is a rare autosomal dominant genetic disorder belonging to the heart-hand syndromes.

Craniosynostosis and dental anomalies is an autosomal recessive syndrome characterized by craniosynostosis, maxillary hypoplasia, and dental anomalies. Dental anomalies seen in this condition include malocclusion, delayed and ectopic tooth eruption, and/or supernumerary teeth. Syndactyly, clinodactyly, and other digit anomalies may also be present.

Craniosynostosis-Dandy-Walker malformation-hydrocephalus syndrome is an autosomal dominant syndrome characterized by sagittal craniosynostosis (scaphocephaly), Dandy-Walker malformation, hydrocephalus, and craniofacial dysmorphisms including hypertelorism, micrognathia, and positional ear deformities.
