
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
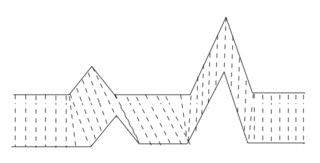
In time series analysis, dynamic time warping (DTW) is an algorithm for measuring similarity between two temporal sequences, which may vary in speed. For instance, similarities in walking could be detected using DTW, even if one person was walking faster than the other, or if there were accelerations and decelerations during the course of an observation. DTW has been applied to temporal sequences of video, audio, and graphics data — indeed, any data that can be turned into a one-dimensional sequence can be analyzed with DTW. A well-known application has been automatic speech recognition, to cope with different speaking speeds. Other applications include speaker recognition and online signature recognition. It can also be used in partial shape matching applications.
In bioinformatics and evolutionary biology, a substitution matrix describes the frequency at which a character in a nucleotide sequence or a protein sequence changes to other character states over evolutionary time. The information is often in the form of log odds of finding two specific character states aligned and depends on the assumed number of evolutionary changes or sequence dissimilarity between compared sequences. It is an application of a stochastic matrix. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where they are used to calculate similarity scores between the aligned sequences.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.

The Needleman–Wunsch algorithm is an algorithm used in bioinformatics to align protein or nucleotide sequences. It was one of the first applications of dynamic programming to compare biological sequences. The algorithm was developed by Saul B. Needleman and Christian D. Wunsch and published in 1970. The algorithm essentially divides a large problem into a series of smaller problems, and it uses the solutions to the smaller problems to find an optimal solution to the larger problem. It is also sometimes referred to as the optimal matching algorithm and the global alignment technique. The Needleman–Wunsch algorithm is still widely used for optimal global alignment, particularly when the quality of the global alignment is of the utmost importance. The algorithm assigns a score to every possible alignment, and the purpose of the algorithm is to find all possible alignments having the highest score.
A Gap penalty is a method of scoring alignments of two or more sequences. When aligning sequences, introducing gaps in the sequences can allow an alignment algorithm to match more terms than a gap-less alignment can. However, minimizing gaps in an alignment is important to create a useful alignment. Too many gaps can cause an alignment to become meaningless. Gap penalties are used to adjust alignment scores based on the number and length of gaps. The five main types of gap penalties are constant, linear, affine, convex, and profile-based.

The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings of nucleic acid sequences or protein sequences. Instead of looking at the entire sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure.
Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm is also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.

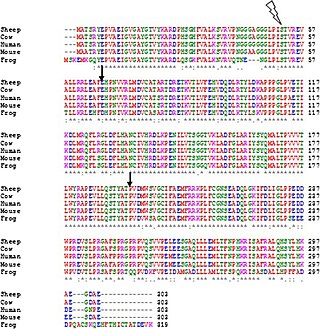
Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
T-Coffee is a multiple sequence alignment software using a progressive approach. It generates a library of pairwise alignments to guide the multiple sequence alignment. It can also combine multiple sequences alignments obtained previously and in the latest versions can use structural information from PDB files (3D-Coffee). It has advanced features to evaluate the quality of the alignments and some capacity for identifying occurrence of motifs (Mocca). It produces alignment in the aln format (Clustal) by default, but can also produce PIR, MSF, and FASTA format. The most common input formats are supported.
Sequential pattern mining is a topic of data mining concerned with finding statistically relevant patterns between data examples where the values are delivered in a sequence. It is usually presumed that the values are discrete, and thus time series mining is closely related, but usually considered a different activity. Sequential pattern mining is a special case of structured data mining.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
In bioinformatics, MAFFT is a program used to create multiple sequence alignments of amino acid or nucleotide sequences. Published in 2002, the first version of MAFFT used an algorithm based on progressive alignment, in which the sequences were clustered with the help of the fast Fourier transform. Subsequent versions of MAFFT have added other algorithms and modes of operation, including options for faster alignment of large numbers of sequences, higher accuracy alignments, alignment of non-coding RNA sequences, and the addition of new sequences to existing alignments.
Warren Richard Gish is the owner of Advanced Biocomputing LLC. He joined Washington University in St. Louis as a junior faculty member in 1994, and was a Research Associate Professor of Genetics from 2002 to 2007.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.
In bioinformatics, a spaced seed is a pattern of relevant and irrelevant positions in a biosequence and a method of approximate string matching that allows for substitutions. They are a straightforward modification to the earliest heuristic-based alignment efforts that allow for minor differences between the sequences of interest. Spaced seeds have been used in homology search., alignment, assembly, and metagenomics. They are usually represented as a sequence of zeroes and ones, where a one indicates relevance and a zero indicates irrelevance at the given position. Some visual representations use pound signs for relevant and dashes or asterisks for irrelevant positions.