Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data. As an interdisciplinary field of science, bioinformatics combines biology, computer science, information engineering, mathematics and statistics to analyze and interpret biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the edit distance cost between strings in a natural language or in financial data.
A phylogenetic tree or evolutionary tree is a branching diagram or "tree" showing the evolutionary relationships among various biological species or other entities—their phylogeny —based upon similarities and differences in their physical or genetic characteristics. All life on Earth is part of a single phylogenetic tree, indicating common ancestry.
Computational biology involves the development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, ecological, behavioral, and social systems. The field is broadly defined and includes foundations in biology, applied mathematics, statistics, biochemistry, chemistry, biophysics, molecular biology, genetics, genomics, computer science and evolution.
Clustal is a series of widely used computer programs used in Bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm are also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.

A phylogenetic network or reticulation is any graph used to visualize evolutionary relationships between nucleotide sequences, genes, chromosomes, genomes, or species. They are employed when reticulation events such as hybridization, horizontal gene transfer, recombination, or gene duplication and loss are believed to be involved. They differ from phylogenetic trees by the explicit modeling of richly linked networks, by means of the addition of hybrid nodes instead of only tree nodes. Phylogenetic trees are a subset of phylogenetic networks. Phylogenetic networks can be inferred and visualised with software such as SplitsTree, the R-package, phangorn, and, more recently, Dendroscope. A standard format for representing phylogenetic networks is a variant of Newick format which is extended to support networks as well as trees.
In computational phylogenetics, tree alignment is a computational problem concerned with producing multiple sequence alignments, or alignments of three or more sequences of DNA, RNA, or protein. Sequences are arranged into a phylogenetic tree, modeling the evolutionary relationships between species or taxa. The edit distances between sequences are calculated for each of the tree's internal vertices, such that the sum of all edit distances within the tree is minimized. Tree alignment can be accomplished using one of several algorithms with various trade-offs between manageable tree size and computational effort.
Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. For example, these techniques have been used to explore the family tree of hominid species and the relationships between specific genes shared by many types of organisms. Traditional phylogenetics relies on morphological data obtained by measuring and quantifying the phenotypic properties of representative organisms, while the more recent field of molecular phylogenetics uses nucleotide sequences encoding genes or amino acid sequences encoding proteins as the basis for classification. Many forms of molecular phylogenetics are closely related to and make extensive use of sequence alignment in constructing and refining phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The phylogenetic trees constructed by computational methods are unlikely to perfectly reproduce the evolutionary tree that represents the historical relationships between the species being analyzed. The historical species tree may also differ from the historical tree of an individual homologous gene shared by those species.

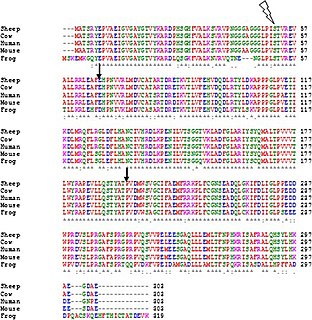
A multiple sequence alignment (MSA) is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.

Intelligent Systems for Molecular Biology (ISMB) is an annual academic conference on the subjects of bioinformatics and computational biology organised by the International Society for Computational Biology (ISCB). The principal focus of the conference is on the development and application of advanced computational methods for biological problems. The conference has been held every year since 1993 and has grown to become one of the largest and most prestigious meetings in these fields, hosting over 2,000 delegates in 2004. From the first meeting, ISMB has been held in locations worldwide; since 2007, meetings have been located in Europe and North America in alternating years. Since 2004, European meetings have been held jointly with the European Conference on Computational Biology (ECCB).

UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.
Quantitative comparative linguistics is the use of quantitative analysis as applied to comparative linguistics.
Bernard M. E. Moret is a Swiss-American computer scientist, an emeritus professor of Computer Science at the École Polytechnique Fédérale de Lausanne in Switzerland. He is known for his work in computational phylogenetics, and in particular for mathematics and methods for computing phylogenetic trees using genome rearrangement events.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.

Ron Shamir is an Israeli professor of computer science known for his work in graph theory and in computational biology. He holds the Raymond and Beverly Sackler Chair in Bioinformatics, and is the founder and head of the Edmond J. Safra Center for Bioinformatics at Tel Aviv University.
Daniel Mier Gusfield is an American computer scientist, Distinguished Professor of Computer Science at the University of California, Davis. Gusfield is known for his research in combinatorial optimization and computational biology.
