| enoyl-CoA, hydratase/3-hydroxyacyl CoA dehydrogenase | |
|---|---|
| Identifiers | |
| Symbol | EHHADH |
| Alt. symbols | ECHD |
| Entrez | 1962 |
| HUGO | 3247 |
| OMIM | 607037 |
| Other data | |
| EC number | 4.2.1.17 |
| Locus | Chr. 3 q26.3-q28 |
EHHADH is a human gene that encodes for a bifunctional enzyme and is one of the four enzymes of the peroxisomal beta-oxidation pathway. Mutations of the gene are a cause of peroxisomal disorders such as Zellweger syndrome. [1]

In biology, a gene is a sequence of nucleotides in DNA or RNA that codes for a molecule that has a function. During gene expression, the DNA is first copied into RNA. The RNA can be directly functional or be the intermediate template for a protein that performs a function. The transmission of genes to an organism's offspring is the basis of the inheritance of phenotypic trait. These genes make up different DNA sequences called genotypes. Genotypes along with environmental and developmental factors determine what the phenotypes will be. Most biological traits are under the influence of polygenes as well as gene–environment interactions. Some genetic traits are instantly visible, such as eye color or number of limbs, and some are not, such as blood type, risk for specific diseases, or the thousands of basic biochemical processes that constitute life.

Enzymes are macromolecular biological catalysts. Enzymes accelerate chemical reactions. The molecules upon which enzymes may act are called substrates and the enzyme converts the substrates into different molecules known as products. Almost all metabolic processes in the cell need enzyme catalysis in order to occur at rates fast enough to sustain life. Metabolic pathways depend upon enzymes to catalyze individual steps. The study of enzymes is called enzymology and a new field of pseudoenzyme analysis has recently grown up, recognising that during evolution, some enzymes have lost the ability to carry out biological catalysis, which is often reflected in their amino acid sequences and unusual 'pseudocatalytic' properties.
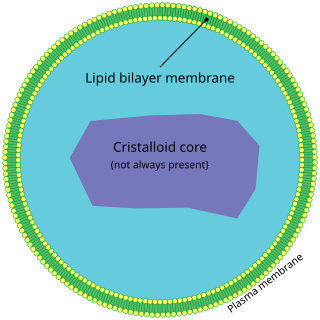
A peroxisome is a type of organelle known as a microbody, found in virtually all eukaryotic cells. They are involved in catabolism of very long chain fatty acids, branched chain fatty acids, D-amino acids, and polyamines, reduction of reactive oxygen species – specifically hydrogen peroxide – and biosynthesis of plasmalogens, i.e., ether phospholipids critical for the normal function of mammalian brains and lungs. They also contain approximately 10% of the total activity of two enzymes in the pentose phosphate pathway, which is important for energy metabolism. It is vigorously debated whether peroxisomes are involved in isoprenoid and cholesterol synthesis in animals. Other known peroxisomal functions include the glyoxylate cycle in germinating seeds ("glyoxysomes"), photorespiration in leaves, glycolysis in trypanosomes ("glycosomes"), and methanol and/or amine oxidation and assimilation in some yeasts.