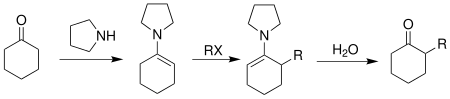
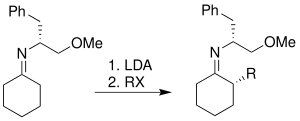
This reaction is a useful technique for asymmetric α-alkylation of ketones and aldehydes, which are common synthetic intermediates for medicinally interesting natural products and other related organic compounds. These natural products include (-)-C10-demethyl arteannuin B, the structural analog of antimalarial artemisinin,[4] the polypropionate metabolite (-)-denticulatin A and B isolated from Siphonaria denticulata,[5]zaragozic acid A, a potent inhibitor of sterol synthesis,[6] and epothilone A and B, which have been proven to be very effective anticancer drugs.[7]
History
Regioselective and stereoselective formation of carbon-carbon bonds adjacent to carbonyl group is an important procedure in organic chemistry. Alkylation reaction of enolates has been the main focus of the field. Both A. G. Myers and D. A. Evans developed asymmetric alkylation reactions for enolates.[8][9]
Myer's and Evans' Asymmetric Alkylation
The apparent shortcoming for enolate alkylation reactions is over-alkylation, even if the amount of base added for enolization as well as the reaction temperature are carefully controlled. The ketene formation during the deprotonation process for substrates possessing Evans' oxazolidinone is also a main side reaction for the related alkylation reactions. Development in the field of enamine chemistry and the utilization of imine derivatives of enolates managed to provide an alternative for enolate alkylation reactions.
In 1976, Meyers reported the first alkylation reaction of metallated azaenolates of hydrazones with an acyclic amino acid-based auxiliary. Compared with the free carbonyl compounds and the chiral enamine species reported previously, the hydrazones exhibit higher reactivity, regioselectivity and stereoselectivity.[11]
Hydrazone alkylation developed by Meyer.
The combination of cyclic amino acid derivatives (SAMP and RAMP) and the powerful hydrazone techniques were pioneered by E. J. Corey and D. Enders in 1976, and were independently developed by D. Enders later. Both SAMP and RAMP are synthesized from amino acids. The detailed synthesis of these two auxiliaries are shown below.[12][13]
EndersSAMP 2
Mechanism
The Enders SAMP/RAMP hydrazone alkylation begins with the synthesis of the hydrazone from a N,N-dialkylhydrazine and a ketone or aldehyde[14]
The hydrazone is then deprotonated on the α-carbon position by a strong base, such as lithium diisopropylamide (LDA), leading to the formation of a resonance stabilized anion - an azaenolate. This anion is a very good nucleophile and readily attacks electrophiles, such as alkyl halides, to generate alkylated hydrazones with simultaneous creation of a new chiral center at the α-carbon.
Mechanism of hydrazone alkylation
The stereochemistry of this reaction is discussed in detail in next section.
Stereochemistry
Stereochemistry of the azaenolate
After the deprotonation, the hydrazone turns into an azaenolate with lithium cation chelating both the nitrogen and oxygen. There are two possible options for lithium chelation. One is that lithium is antiperiplanar to the C=C bond (blue colored), leading to the conformation of ZC-N; the other one is that lithium and the C=C bond are at the same side of the C-N bond (red colored), leading to the EC-N conformer. There are also two available orientations for the chelating nitrogen and R2 group, being either EC=C or ZC=C. This leads to four possible azaenolate intermediates (A, B, C and D) for the Enders' SAMP/RAMP hydrazone alkylation reaction.
Stereoselectivity of the generation of the azaenolates
Experiments and calculations[2][15][16] show that one specific stereoisomer of the azaenolate is favored over the other three possible candidates. Therefore, although four isomers are possible for the azaenolate, only the one (azaenolate A) with the stereochemistry of its C=C double bonds being E and that of its C-N bond being Z stereochemistry is dominant (EC=CZC-N) for both cyclic and acyclic ketones.[17]
Stereochemistry of alkylation
The favored azaenolate is the dominant starting molecule for the subsequent alkylation reaction. There are two possible faces of accessing for any electrophile to react with. The steric interaction between the pyrrolidine ring and the electrophilic reagent hinders the attack of the electrophile from the top face. On the contrary, when the electrophile attacks from the bottom face, such unfavorable interaction does not exist. Therefore, the electrophilic attack proceeds from the sterically more accessible face.[18]
Stereoselectivity for the alkylation step of the Enders' reaction
Variants
The chelation of lithiumcation with the methoxy group is one of the most important features of the transition state for Enders' hydrazone alkylation reaction. It is necessary to have this chelation effect to achieve high stereoselectivity. The development and modification of Enders' hydrazone alkylation reaction mainly focus on the addition of more steric hindrance on the pyrrolidine rings of both SAMP and RAMP, while preserving the methoxy group for lithium chelation.
The most famous four variants of SAMP and RAMP are SADP, SAEP, SAPP and RAMBO,[19][20] whose structures are shown below.
Variants of SAMP/RAMP with bulkier groups attached
In 2011, several N-amino cyclic carbamates were synthesized and studied for asymmetric hydrazone alkylation reactions.[21] Both the stereochemistry and regioselectivity of the reactions turned out to be very promising. These new compounds consist of a new class of chiral auxiliary based on the carbamate structure and, therefore, no longer belong to the family of SAMP and RAMP. But they do provide very powerful alternatives to the traditional pyrrolidine systems.
N-Amino cyclic carbamates
Auxiliary release
Hydrazones are usually very stable towards solvolysis, and conversion to the ketone can require vigorous conditions. Also, aldehydic hydrazones often instead disproportionate to a nitrile and amine.[22]
Two principal workup environments are common: oxidation and solvolysis. Reductive conversions are possible with low-valenttransition metals, but remained relatively unstudied as of 2000[update].[22]
Indeed, a wide variety of acids promote hydrolysis. Bismuth trichloride cleaves arbitrary hydrazones in a microwave. Oxalic acid abstracts hydrazine from ketonic hydrazones; the oxalate adduct then decomposes to the original auxiliary in aqueous base. Silica gel hydrolyzes exquisitely acid-sensitive substrates, but is too weak to affect ketonic hydrazones adjacent to a primary carbon. Ketonic hydrazones adjacent to a secondary or tertiary carbon hydrolyze in the presence of catalyticcupricsalts; that procedure also preserves substrates disturbed by oxidants or strong acids.[22]
Hydrazone carbamates are cleaved much more readily than their parent hydrazones: para-toluenesulfonic acid affords the corresponding ketones in near-quantitative yields.[21]
Conditions
Ender's hydrazone alkylation reaction is usually run through a sequence of three steps.[14] The first step should always be the synthesis of the hydrazones. The ketone or aldehyde is mixed with either SAMP or RAMP and allowed to react under argon for 12 hours. The crude hydrazone obtained is purified by distillation or recrystallization. At 0 degree celsius, the hydrazone is transferred into the ether solution of lithium diisopropylamide. Then this mixture is cooled down to -110 degree celsius and is slowly added the alkyl halide. This mixture is then allowed to warm up to room temperature. After 12 hours of reaction at room temperature, the crude alkylated hydrazone is allowed to react with ozone in a Schlenk tube to cleave the C=N bond. After distillation or column chromatography, pure alkylation product can be obtained.
Applications
Synthesis of zaragozic acid A
K. C. Nicolaou and coworkers at Scripps Research Institute generated the chiral hydrazone through Enders' hydrazone alkylation reaction with high stereoselectivity (de > 95%). The subsequent ozonolysis and Wittig reaction led to the side chain fragment of zaragozic acid A, which is a potent medicine for coronary heart disease.[6]
Synthesis of Zaragozic Acid
Synthesis of denticulatin A and B
Ziegler and coworkers reacted an allyliodide with the azaenolate to generate a chiral hydrocarbon chain. To avoid loss of the enantiomeric purity of the product, the authors used cupric acetate to regenerate the carbonyl group, obtaining only moderate yield for the cleavage of C=N bond but good enantioselectivity (ee = 89%). The ketone was transformed after several steps into denticulatin A and B - polypropionate metabolites isolated from Siphonaria Denticulata.[5]
Synthesis of Denticulatin A&B
Synthesis of the derivative of arteannuin
(-)-C10-demethyl arteannuin B is a structural analog of the antimalarialartemisinin. It exhibits potent antimalarial activity even against a drug-resistant strain. Little and coworkers obtained the alkylated hydrazone in diastereomerically pure form (de > 95%) through the Enders' alkylation reaction. This intermediate was then elaborated into (-)-C10-demethyl arteannuin B.[4]
Synthesis of Arteannuin B
Synthesis of epothilone A
Epothilone A and B are reported to be highly effective anticancer drugs. Several of their structural derivatives show very promising inhibition against breast cancer with only mild side effect and some of them are now under trials. In 1997, K. C. Nicolaou and coworkers reported the first total synthesis of both Epothilone A and B. Ender's alkylation reaction was utilized at the very beginning of the synthesis to install the stereogenic center at C8. The reaction proceeded with both high yield and high diastereoselectivity.[7]
↑ Corey, E. J.; Enders, D. (1976). "Applications of N,N-dimethylhydrazones to synthesis. Use in efficient, positionally and stereochemically selective C-C bond formation; oxidative hydrolysis to carbonyl compounds". Tetrahedron Letters. 17 (1): 3–6. doi:10.1016/s0040-4039(00)71307-4.
1 2 Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington, MA: Elsevier Academic Press. pp.150–151. ISBN0-12-369483-3.
↑ Job, A.; Janeck, C. F.; Bettray, W.; Peters, R.; Enders, D. (2002). "The SAMP-/RAMP-hydrazone methodology in asymmetric synthesis". Tetrahedron. 58 (12): 2253–2329. doi:10.1016/s0040-4020(02)00080-7.
1 2 Nicolaou, K. C.; Ninkovic, S.; Sarabia, F.; Vourloumis, D.; He, Y.; Vallberg, H.; Finlay, M. R. V.; Yang, Z. (1997). "Total Syntheses of Epothilones A and B via a Macrolactonization-Based Strategy". Journal of the American Chemical Society. 119 (34): 7974–7991. doi:10.1021/ja971110h.
↑ Myers, A. G., Yang, B. H., Chen, H., McKinstry, L. Kopecky, D. J., Gleason, J. L. (1997). "Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones". Journal of the American Chemical Society. 119 (28): 6496–6511. doi:10.1021/ja970402f.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ Evans, D. A.; Ennis, M. D.; Mathre, D. J. (1982). "Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives". Journal of the American Chemical Society. 104 (6): 1737–1739. doi:10.1021/ja00370a050.
↑ Enders, D.; Eichenauer, H.; Pieter, R. (1979). "Enantioselektive Synthese von (-)-(R)-und(+)-(S)-[6]-Gingerol-Gewürzprinzip des Ingwers". Chemische Berichte. 112 (11): 3703–3714. doi:10.1002/cber.19791121118.
↑ Enders, D.; Eichenauer, H. (1979). "Asymmetrische Synthesen via metallierte chirale Hydrazone. Enantioselektive Alkylierung von cyclischen Ketonen und Aldehyden". Chemische Berichte. 112 (8): 2933–2960. doi:10.1002/cber.19791120820.
1 2 Enders, D., Kipphardt, H., Fey1, P. (1987). "Asymmetric Synthesis Using the SAMP-/RAMP- Hydrazone Method: (S)-(+)-4-Methyl-3-Heptanoe". Organic Syntheses. 65: 183. doi:10.15227/orgsyn.065.0183.{{cite journal}}: CS1 maint: multiple names: authors list (link) CS1 maint: numeric names: authors list (link)
↑ Ahlbrecht, H.; Düber, E. O.; Enders, D.; Eichenauer, H.; Weuster, P. (1978). "NMR-spektroskopische untersuchungen an deprotonierten iminen und hydrazonen". Tetrahedron Letters. 19 (39): 3691–3694. doi:10.1016/s0040-4039(01)95032-4.
↑ Enders, D.; Baus, U. (1983). "Asymmetrische Synthese beider Enantiomere von (E)-4,6-Dimethyl-6-octen-3-on – Abwehrsubstanz der Weberknechte Leiobunum vittatum und L. calcar (Opiliones)". Liebigs Annalen der Chemie. 1983 (8): 1439–1445. doi:10.1002/jlac.198319830816.
↑ Enders, D.; Bachstädter, G.; Kremer, K. A. M.; Marsch, M.; Harms, K.; Boche, G. (1988). "Structure of a Chiral Lithium Azaenolate: Monomeric, Intramolecular Chelated Lithio-2-acetylnaphthalene-SAMP-hydrazone". Angewandte Chemie International Edition in English. 27 (11): 1522–1524. doi:10.1002/anie.198815221.
↑ Bauer, W.; Seebach, D. (1984). "Bestimmung des Aggregationsgrads lithiumorganischer Verbindungen durch Kryoskopie in Tetrahydrofuran". Helvetica Chimica Acta. 67 (7): 1972–1988. doi:10.1002/hlca.19840670736.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.