
Inorganic chemistry deals with synthesis and behavior of inorganic and organometallic compounds. This field covers chemical compounds that are not carbon-based, which are the subjects of organic chemistry. The distinction between the two disciplines is far from absolute, as there is much overlap in the subdiscipline of organometallic chemistry. It has applications in every aspect of the chemical industry, including catalysis, materials science, pigments, surfactants, coatings, medications, fuels, and agriculture.
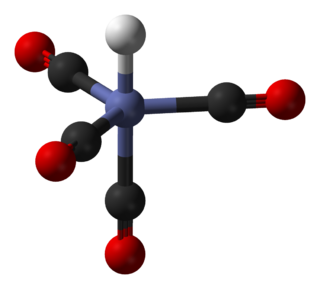
In coordination chemistry, a ligand is an ion or molecule with a functional group that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs, often through Lewis bases. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".
Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.

In biochemistry and molecular biology, a binding site is a region on a macromolecule such as a protein that binds to another molecule with specificity. The binding partner of the macromolecule is often referred to as a ligand. Ligands may include other proteins, enzyme substrates, second messengers, hormones, or allosteric modulators. The binding event is often, but not always, accompanied by a conformational change that alters the protein's function. Binding to protein binding sites is most often reversible, but can also be covalent reversible or irreversible.
A hormone receptor is a receptor molecule that binds to a specific hormone. Hormone receptors are a wide family of proteins made up of receptors for thyroid and steroid hormones, retinoids and Vitamin D, and a variety of other receptors for various ligands, such as fatty acids and prostaglandins. Hormone receptors are of mainly two classes. Receptors for peptide hormones tend to be cell surface receptors built into the plasma membrane of cells and are thus referred to as trans membrane receptors. An example of this is Actrapid. Receptors for steroid hormones are usually found within the protoplasm and are referred to as intracellular or nuclear receptors, such as testosterone. Upon hormone binding, the receptor can initiate multiple signaling pathways, which ultimately leads to changes in the behavior of the target cells.
A substitution reaction is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group. Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.
Oxidative addition and reductive elimination are two important and related classes of reactions in organometallic chemistry. Oxidative addition is a process that increases both the oxidation state and coordination number of a metal centre. Oxidative addition is often a step in catalytic cycles, in conjunction with its reverse reaction, reductive elimination.

In chemistry, octahedral molecular geometry, also called square bipyramidal, describes the shape of compounds with six atoms or groups of atoms or ligands symmetrically arranged around a central atom, defining the vertices of an octahedron. The octahedron has eight faces, hence the prefix octa. The octahedron is one of the Platonic solids, although octahedral molecules typically have an atom in their centre and no bonds between the ligand atoms. A perfect octahedron belongs to the point group Oh. Examples of octahedral compounds are sulfur hexafluoride SF6 and molybdenum hexacarbonyl Mo(CO)6. The term "octahedral" is used somewhat loosely by chemists, focusing on the geometry of the bonds to the central atom and not considering differences among the ligands themselves. For example, [Co(NH3)6]3+, which is not octahedral in the mathematical sense due to the orientation of the N−H bonds, is referred to as octahedral.

The Jacobsen epoxidation, sometimes also referred to as Jacobsen-Katsuki epoxidation is a chemical reaction which allows enantioselective epoxidation of unfunctionalized alkyl- and aryl- substituted alkenes. It is complementary to the Sharpless epoxidation (used to form epoxides from the double bond in allylic alcohols). The Jacobsen epoxidation gains its stereoselectivity from a C2 symmetric manganese(III) salen-like ligand, which is used in catalytic amounts. The manganese atom transfers an oxygen atom from chlorine bleach or similar oxidant. The reaction takes its name from its inventor, Eric Jacobsen, with Tsutomu Katsuki sometimes being included. Chiral-directing catalysts are useful to organic chemists trying to control the stereochemistry of biologically active compounds and develop enantiopure drugs.
The transforming growth factor beta (TGFB) signaling pathway is involved in many cellular processes in both the adult organism and the developing embryo including cell growth, cell differentiation, cell migration, apoptosis, cellular homeostasis and other cellular functions. The TGFB signaling pathways are conserved. In spite of the wide range of cellular processes that the TGFβ signaling pathway regulates, the process is relatively simple. TGFβ superfamily ligands bind to a type II receptor, which recruits and phosphorylates a type I receptor. The type I receptor then phosphorylates receptor-regulated SMADs (R-SMADs) which can now bind the coSMAD SMAD4. R-SMAD/coSMAD complexes accumulate in the nucleus where they act as transcription factors and participate in the regulation of target gene expression.
In organometallic chemistry, a transition metal indenyl complex is a coordination compound that contains one or more indenyl ligands. The indenyl ligand is formally the anion derived from deprotonation of indene. The η5-indenyl ligand is related to the η5cyclopentadienyl anion (Cp), thus indenyl analogues of many cyclopentadienyl complexes are known. Indenyl ligands lack the 5-fold symmetry of Cp, so they exhibit more complicated geometries. Furthermore, some indenyl complexes also exist with only η3-bonding mode. The η5- and η3-bonding modes sometimes interconvert.
Associative substitution describes a pathway by which compounds interchange ligands. The terminology is typically applied to organometallic and coordination complexes, but resembles the Sn2 mechanism in organic chemistry. The opposite pathway is dissociative substitution, being analogous to the Sn1 pathway. Intermediate pathways exist between the pure associative and pure dissociative pathways, these are called interchange mechanisms.
In chemistry, dissociative substitution describes a reaction pathway by which compounds interchange ligands. The term is typically applied to coordination and organometallic complexes, but resembles the SN1 mechanism in organic chemistry. This pathway can be well described by the cis effect, or the labilization of CO ligands in the cis position. The opposite pathway is associative substitution, being analogous to SN2 pathway. Pathways that are intermediate between the pure dissociative and pure associative pathways are called interchange mechanisms.

Organomolybdenum chemistry is the chemistry of chemical compounds with Mo-C bonds. The heavier group 6 elements molybdenum and tungsten form organometallic compounds similar to those in organochromium chemistry but higher oxidation states tend to be more common.
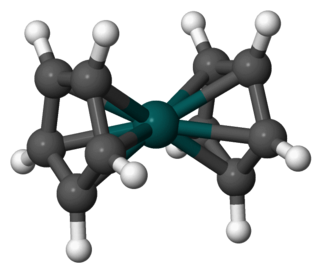
Rhodocene is a chemical compound with the formula [Rh(C5H5)2]. Each molecule contains an atom of rhodium bound between two planar aromatic systems of five carbon atoms known as cyclopentadienyl rings in a sandwich arrangement. It is an organometallic compound as it has (haptic) covalent rhodium–carbon bonds. The [Rh(C5H5)2] radical is found above 150 °C (302 °F) or when trapped by cooling to liquid nitrogen temperatures (−196 °C [−321 °F]). At room temperature, pairs of these radicals join via their cyclopentadienyl rings to form a dimer, a yellow solid.

NKG2D is an activating receptor (transmembrane protein) belonging to the NKG2 family of C-type lectin-like receptors. NKG2D is encoded by KLRK1 (killer cell lectin like receptor K1) gene which is located in the NK-gene complex (NKC) situated on chromosome 6 in mice and chromosome 12 in humans. In mice, it is expressed by NK cells, NK1.1+ T cells, γδ T cells, activated CD8+ αβ T cells and activated macrophages. In humans, it is expressed by NK cells, γδ T cells and CD8+ αβ T cells. NKG2D recognizes induced-self proteins from MIC and RAET1/ULBP families which appear on the surface of stressed, malignant transformed, and infected cells.
In organometallic chemistry, a transition metal alkene complex is a coordination compound containing one or more alkene ligands. The inventory is large. Such compounds are intermediates in many catalytic reactions that convert alkenes to other organic products.
Gallium monoiodide is an inorganic gallium compound with the formula GaI or Ga4I4. It is a pale green solid and mixed valent gallium compound, which can contain gallium in the 0, +1, +2, and +3 oxidation states. It is used as a pathway for many gallium-based products. Unlike the gallium(I) halides first crystallographically characterized, gallium monoiodide has a more facile synthesis allowing a synthetic route to many low-valent gallium compounds.

Chlorine-free germanium processing are methods of germanium activation to form useful germanium precursors in a more energy efficient and environmentally friendly way compared to traditional synthetic routes. Germanium tetrachloride is a valuable intermediate for the synthesis of many germanium complexes. Normal synthesis of it involves an energy-intensive dehydration of germanium oxide, , with hydrogen chloride, Due to the environmental and safety impact of non-recyclable, high energy reactions with , an alternative synthesis of a shelf-stable germanium intermediate precursor without chlorine is of interest. In 2017, a synthesis of organogermanes, without using chloride species was reported, allowing for a much more environmentally friendly and low energy synthesis using , , and even selectively activating germanium in the presence of zinc oxide, resulting in products that are bench stable and solid.