
Prader–Willi syndrome (PWS) is a genetic disorder caused by a loss of function of specific genes on chromosome 15. In newborns, symptoms include weak muscles, poor feeding, and slow development. Beginning in childhood, those affected become constantly hungry, which often leads to obesity and type 2 diabetes. Mild to moderate intellectual impairment and behavioral problems are also typical of the disorder. Often, affected individuals have a narrow forehead, small hands and feet, short height, light skin and hair. Most are unable to have children.

Ubiquitin-protein ligase E3A (UBE3A) also known as E6AP ubiquitin-protein ligase (E6AP) is an enzyme that in humans is encoded by the UBE3A gene. This enzyme is involved in targeting proteins for degradation within cells.

Chromosome 15 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 15 spans about 101 million base pairs and represents between 3% and 3.5% of the total DNA in cells.

Gamma-aminobutyric acid receptor subunit beta-3 is a protein that in humans is encoded by the GABRB3 gene. It is located within the 15q12 region in the human genome and spans 250kb. This gene includes 10 exons within its coding region. Due to alternative splicing, the gene codes for many protein isoforms, all being subunits in the GABAA receptor, a ligand-gated ion channel. The beta-3 subunit is expressed at different levels within the cerebral cortex, hippocampus, cerebellum, thalamus, olivary body and piriform cortex of the brain at different points of development and maturity. GABRB3 deficiencies are implicated in many human neurodevelopmental disorders and syndromes such as Angelman syndrome, Prader-Willi syndrome, nonsyndromic orofacial clefts, epilepsy and autism. The effects of methaqualone and etomidate are mediated through GABBR3 positive allosteric modulation.

Small nuclear ribonucleoprotein-associated protein N is a protein that in humans is encoded by the SNRPN gene.
Necdin is a protein that in humans is encoded by the NDN gene.
SNRPN upstream reading frame protein is a protein that in humans is encoded by the SNURF gene.
Non-imprinted in Prader-Willi/Angelman syndrome region protein 1 is a protein that in humans is encoded by the NIPA1 gene. This gene encodes a potential transmembrane protein which functions either as a receptor or transporter molecule, possibly as a magnesium transporter. This protein is thought to play a role in nervous system development and maintenance. Alternative splice variants have been described, but their biological nature has not been determined. Mutations in this gene have been associated with the human genetic disease autosomal dominant spastic paraplegia 6.
Prader-Willi/Angelman region-1, also known as PWAR1, is an exon of the lncRNA Small nucleolar RNA host gene 14 (SNHG14).
Gamma-tubulin complex component 5 is a protein that in humans is encoded by the TUBGCP5 gene. It is part of the gamma tubulin complex, which required for microtubule nucleation at the centrosome.
Probable phospholipid-transporting ATPase VA also known as ATPase class V type 10A (ATP10A) or aminophospholipid translocase VA is an enzyme that in humans is encoded by the ATP10A gene.
Non-imprinted in Prader-Willi/Angelman syndrome region protein 2 is a protein that in humans is encoded by the NIPA2 gene.
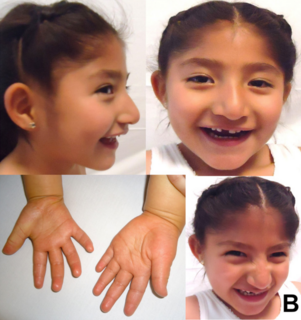
Angelman syndrome or Angelman's syndrome (AS) is a genetic disorder that mainly affects the nervous system. Symptoms include a small head and a specific facial appearance, severe intellectual disability, developmental disability, limited to no functional speech, balance and movement problems, seizures, and sleep problems. Children usually have a happy personality and have a particular interest in water. The symptoms generally become noticeable by one year of age.
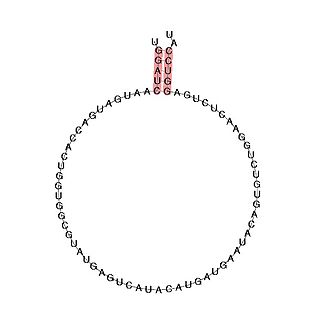
Small nucleolar RNA SNORD113 is a small nucleolar RNA molecule which is located in the imprinted human 14q32 locus and may play a role in the evolution and/or mechanism of the epigenetic imprinting process.
In molecular biology, Maternally expressed 8, also known as MEG8 or Rian, is a long non-coding RNA. It is an imprinted gene, which is maternally expressed. It is expressed in the nucleus and is preferentially expressed in skeletal muscle.

UBE3A-ATS/Ube3a-ATS (human/mouse), otherwise known as ubiquitin ligase E3A-ATS, is the name for the antisense DNA strand that is transcribed as part of a larger transcript called LNCAT at the Ube3a locus. The Ube3a locus is imprinted and in the central nervous system expressed only from the maternal allele. Silencing of Ube3a on the paternal allele is thought to occur through the Ube3a-ATS part of LNCAT, since non-coding antisense transcripts are often found at imprinted loci. The deletion and/or mutation of Ube3a on the maternal chromosome causes Angelman Syndrome (AS) and Ube3a-ATS may prove to be an important aspect in finding a therapy for this disease. While in patients with AS the maternal Ube3a allele is inactive, the paternal allele is intact but epigenetically silenced. If unsilenced, the paternal allele could be a source of active Ube3a protein in AS patients. Therefore, understanding the mechanisms of how Ube3a-ATS might be involved in silencing the paternal Ube3a may lead to new therapies for AS. This possibility has been demonstrated by a recent study where the drug topotecan, administered to mice suffering from AS, activated expression of the paternal Ube3a gene by lowering the transcription of Ube3a-ATS.

Chromosomal deletion syndromes result from deletion of parts of chromosomes. Depending on the location, size, and whom the deletion is inherited from, there are a few known different variations of chromosome deletions. Chromosomal deletion syndromes typically involve larger deletions that are visible using karyotyping techniques. Smaller deletions result in Microdeletion syndrome, which are detected using fluorescence in situ hybridization (FISH)
Microdeletion syndrome is a syndrome caused by a chromosomal deletion smaller than 5 million base pairs spanning several genes that is too small to be detected by conventional cytogenetic methods or high resolution karyotyping. Detection is done by fluorescence in situ hybridization (FISH). Larger chromosomal deletion syndromes are detectable using karyotyping techniques.
Makorin ring finger protein 3 is a protein that in humans is encoded by the MKRN3 gene.
Intragenomic and intrauterine conflicts in humans arise between mothers and their offspring. Parental investment theory states that parents and their offspring will often be in conflict over the optimal amount of investment that the parent should provide. This is because the best interests of the parent do not always match the best interests of the offspring. Maternal-infant conflict is of interest due to the intensity of maternal investment in her offspring. In humans, mothers often invest years of care into their children due to the long developmental period before children become self-sufficient.