Related Research Articles

In molecular biology and biotechnology, a fluorescent tag, also known as a fluorescent label or fluorescent probe, is a molecule that is attached chemically to aid in the detection of a biomolecule such as a protein, antibody, or amino acid. Generally, fluorescent tagging, or labeling, uses a reactive derivative of a fluorescent molecule known as a fluorophore. The fluorophore selectively binds to a specific region or functional group on the target molecule and can be attached chemically or biologically. Various labeling techniques such as enzymatic labeling, protein labeling, and genetic labeling are widely utilized. Ethidium bromide, fluorescein and green fluorescent protein are common tags. The most commonly labelled molecules are antibodies, proteins, amino acids and peptides which are then used as specific probes for detection of a particular target.
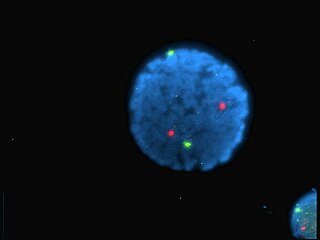
A fluorophore is a fluorescent chemical compound that can re-emit light upon light excitation. Fluorophores typically contain several combined aromatic groups, or planar or cyclic molecules with several π bonds.
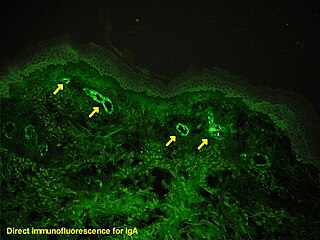
Immunofluorescence is a technique used for light microscopy with a fluorescence microscope and is used primarily on biological samples. This technique uses the specificity of antibodies to their antigen to target fluorescent dyes to specific biomolecule targets within a cell, and therefore allows visualization of the distribution of the target molecule through the sample. The specific region an antibody recognizes on an antigen is called an epitope. There have been efforts in epitope mapping since many antibodies can bind the same epitope and levels of binding between antibodies that recognize the same epitope can vary. Additionally, the binding of the fluorophore to the antibody itself cannot interfere with the immunological specificity of the antibody or the binding capacity of its antigen. Immunofluorescence is a widely used example of immunostaining and is a specific example of immunohistochemistry. This technique primarily makes use of fluorophores to visualise the location of the antibodies.
In biochemistry, biotinylation is the process of covalently attaching biotin to a protein, nucleic acid or other molecule. Biotinylation is rapid, specific and is unlikely to disturb the natural function of the molecule due to the small size of biotin. Biotin binds to streptavidin and avidin with an extremely high affinity, fast on-rate, and high specificity, and these interactions are exploited in many areas of biotechnology to isolate biotinylated molecules of interest. Biotin-binding to streptavidin and avidin is resistant to extremes of heat, pH and proteolysis, making capture of biotinylated molecules possible in a wide variety of environments. Also, multiple biotin molecules can be conjugated to a protein of interest, which allows binding of multiple streptavidin, avidin or neutravidin protein molecules and increases the sensitivity of detection of the protein of interest. There is a large number of biotinylation reagents available that exploit the wide range of possible labelling methods. Due to the strong affinity between biotin and streptavidin, the purification of biotinylated proteins has been a widely used approach to identify protein-protein interactions and post-translational events such as ubiquitylation in molecular biology.

Förster resonance energy transfer (FRET), fluorescence resonance energy transfer, resonance energy transfer (RET) or electronic energy transfer (EET) is a mechanism describing energy transfer between two light-sensitive molecules (chromophores). A donor chromophore, initially in its electronic excited state, may transfer energy to an acceptor chromophore through nonradiative dipole–dipole coupling. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making FRET extremely sensitive to small changes in distance.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.
Chemical biology is a scientific discipline between the fields of chemistry and biology. The discipline involves the application of chemical techniques, analysis, and often small molecules produced through synthetic chemistry, to the study and manipulation of biological systems. In contrast to biochemistry, which involves the study of the chemistry of biomolecules and regulation of biochemical pathways within and between cells, chemical biology deals with chemistry applied to biology.

Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only particular parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.
In chemical synthesis, click chemistry is a class of simple, atom-economy reactions commonly used for joining two molecular entities of choice. Click chemistry is not a single specific reaction, but describes a way of generating products that follow examples in nature, which also generates substances by joining small modular units. In many applications, click reactions join a biomolecule and a reporter molecule. Click chemistry is not limited to biological conditions: the concept of a "click" reaction has been used in chemoproteomic, pharmacological, and various biomimetic applications. However, they have been made notably useful in the detection, localization and qualification of biomolecules.

Molecular imaging is a field of medical imaging that focuses on imaging molecules of medical interest within living patients. This is in contrast to conventional methods for obtaining molecular information from preserved tissue samples, such as histology. Molecules of interest may be either ones produced naturally by the body, or synthetic molecules produced in a laboratory and injected into a patient by a doctor. The most common example of molecular imaging used clinically today is to inject a contrast agent into a patient's bloodstream and to use an imaging modality to track its movement in the body. Molecular imaging originated from the field of radiology from a need to better understand fundamental molecular processes inside organisms in a noninvasive manner.
Fluorescence Loss in Photobleaching (FLIP) is a fluorescence microscopy technique used to examine movement of molecules inside cells and membranes. A cell membrane is typically labeled with a fluorescent dye to allow for observation. A specific area of this labeled section is then bleached several times using the beam of a confocal laser scanning microscope. After each imaging scan, bleaching occurs again. This occurs several times, to ensure that all accessible fluorophores are bleached since unbleached fluorophores are exchanged for bleached fluorophores, causing movement through the cell membrane. The amount of fluorescence from that region is then measured over a period of time to determine the results of the photobleaching on the cell as a whole.

Activity-based proteomics, or activity-based protein profiling (ABPP) is a functional proteomic technology that uses chemical probes that react with mechanistically related classes of enzymes.
Alice Yen-Ping Ting is Taiwanese-born American chemist. She is a professor of Genetics, of Biology, and by courtesy, of Chemistry at Stanford University. She is also a Chan Zuckerberg Biohub investigator.
EosFP is a photoactivatable green to red fluorescent protein. Its green fluorescence (516 nm) switches to red (581 nm) upon UV irradiation of ~390 nm due to a photo-induced modification resulting from a break in the peptide backbone near the chromophore. Eos was first discovered as a tetrameric protein in the stony coral Lobophyllia hemprichii. Like other fluorescent proteins, Eos allows for applications such as the tracking of fusion proteins, multicolour labelling and tracking of cell movement. Several variants of Eos have been engineered for use in specific study systems including mEos2, mEos4 and CaMPARI.

Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence. Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.
Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit, which is due to the diffraction of light. Super-resolution imaging techniques rely on the near-field or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution or detector-based pixel reassignment, the 4Pi microscope, and structured-illumination microscopy technologies such as SIM and SMI.
Photo-activated localization microscopy and stochastic optical reconstruction microscopy (STORM) are widefield fluorescence microscopy imaging methods that allow obtaining images with a resolution beyond the diffraction limit. The methods were proposed in 2006 in the wake of a general emergence of optical super-resolution microscopy methods, and were featured as Methods of the Year for 2008 by the Nature Methods journal. The development of PALM as a targeted biophysical imaging method was largely prompted by the discovery of new species and the engineering of mutants of fluorescent proteins displaying a controllable photochromism, such as photo-activatible GFP. However, the concomitant development of STORM, sharing the same fundamental principle, originally made use of paired cyanine dyes. One molecule of the pair, when excited near its absorption maximum, serves to reactivate the other molecule to the fluorescent state.

SNAP-tag® is a self-labeling protein tag commercially available in various expression vectors. SNAP-tag is a 182 residues polypeptide that can be fused to any protein of interest and further specifically and covalently tagged with a suitable ligand, such as a fluorescent dye. Since its introduction, SNAP-tag has found numerous applications in biochemistry and for the investigation of the function and localisation of proteins and enzymes in living cells. Compared to the current standard labelling methods used in fluorescence microscopy, the use of SNAP-tag presents significant advantages. SNAP-tag® is a registered trademark of New England Biolabs, Inc.

Small ultra red fluorescent protein (smURFP) is a class of far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein, α-allophycocyanin. Native α-allophycocyanin requires an exogenous protein, known as a lyase, to attach the chromophore, phycocyanobilin. Phycocyanobilin is not present in mammalian cells. smURFP was evolved to covalently attach phycocyanobilin without a lyase and fluoresce, covalently attach biliverdin and fluoresce, blue-shift fluorescence to match the organic fluorophore, Cy5, and not inhibit E. coli growth. smURFP was found after 12 rounds of random mutagenesis and manually screening 10,000,000 bacterial colonies.
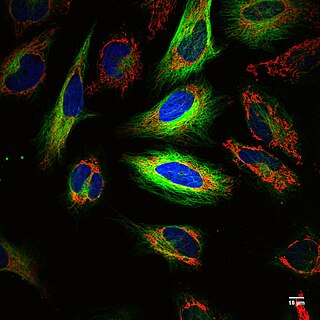
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.
References
- 1 2 3 4 5 6 7 Uttamapinant C, Sanchez MI, Liu DS, Yao JZ, Ting AY (August 2013). "Site-specific protein labeling using PRIME and chelation-assisted click chemistry". Nat Protoc. 8 (8): 1620–34. doi:10.1038/nprot.2013.096. PMC 4892701 . PMID 23887180.
- ↑ Tschesche H, ed. (2011). Methods in protein biochemistry. Berlin: De Gruyter. ISBN 978-3-11-025236-1.
- 1 2 Soh N (19 February 2008). "Selective Chemical Labeling of Proteins with Small Fluorescent Molecules Based on Metal-Chelation Methodology". Sensors. 8 (2): 1004–1024. Bibcode:2008Senso...8.1004S. doi: 10.3390/s8021004 . PMC 3927527 . PMID 27879749.
- 1 2 3 Yao JZ, Uttamapinant C, Poloukhtine A, Baskin JM, Codelli JA, Sletten EM, Bertozzi CR, Popik VV, Ting AY (2012). "Fluorophore targeting to cellular proteins via enzyme-mediated azide ligation and strain-promoted cycloaddition". J. Am. Chem. Soc. 134 (8): 3720–8. doi:10.1021/ja208090p. PMC 3306817 . PMID 22239252.
- ↑ Demchenko AP, Brouwer AM, ed. (2011). Advanced fluorescence reporters in chemistry and biology. Heidelberg: Springer. ISBN 978-3-642-18034-7.
- ↑ Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM, Ting AY (2012). "Diels-Alder cycloaddition for fluorophore targeting to specific proteins inside living cells". J. Am. Chem. Soc. 134 (2): 792–5. doi:10.1021/ja209325n. PMC 3381951 . PMID 22176354.
- ↑ Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY (2012). "Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling". Angew. Chem. Int. Ed. Engl. 51 (24): 5852–6. doi:10.1002/anie.201108181. PMC 3517120 . PMID 22555882.
- ↑ Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY (2010). "A fluorophore ligase for site-specific protein labeling inside living cells". Proc Natl Acad Sci U S A. 107 (24): 10914–9. doi: 10.1073/pnas.0914067107 . PMC 2890758 . PMID 20534555.
- ↑ Liu DS, Nivon LG, Richter F, Goldman PJ, Deerinck TJ, Yao JZ, Phipps WS, Ellisman MH, Drennan CL, Baker D, Ting AY (2014). "Computational design of a red fluorophore ligase for site-specific protein labeling in living cells". Proc Natl Acad Sci U S A. 111 (43): E4551–9. Bibcode:2014PNAS..111E4551L. doi: 10.1073/pnas.1404736111 . PMC 4217414 . PMID 25313043.