
In genetics, complementary DNA (cDNA) is DNA synthesized from a single-stranded RNA template in a reaction catalyzed by the enzyme reverse transcriptase. cDNA is often used to clone eukaryotic genes in prokaryotes. When scientists want to express a specific protein in a cell that does not normally express that protein, they will transfer the cDNA that codes for the protein to the recipient cell. In molecular biology, cDNA is also generated to analyze transcriptomic profiles in bulk tissue, single cells, or single nuclei in assays such as microarrays and RNA-seq.

Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation. It is fundamental to many of the procedures used in genetic testing and research, including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory research for a broad variety of applications including biomedical research and criminal forensics.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable.

A reverse transcriptase (RT) is an enzyme used to generate complementary DNA (cDNA) from an RNA template, a process termed reverse transcription. Reverse transcriptases are used by certain viruses such as HIV and hepatitis B to replicate their genomes, by retrotransposon mobile genetic elements to proliferate within the host genome, and by eukaryotic cells to extend the telomeres at the ends of their linear chromosomes. Contrary to a widely held belief, the process does not violate the flows of genetic information as described by the classical central dogma, as transfers of information from RNA to DNA are explicitly held possible.

Transcription is the process of copying a segment of DNA into RNA. The segments of DNA transcribed into RNA molecules that can encode proteins are said to produce messenger RNA (mRNA). Other segments of DNA are copied into RNA molecules called non-coding RNAs (ncRNAs). Averaged over multiple cell types in a given tissue, the quantity of mRNA is more than 10 times the quantity of ncRNA. The general preponderance of mRNA in cells is valid even though less than 2% of the human genome can be transcribed into mRNA, while at least 80% of mammalian genomic DNA can be actively transcribed, with the majority of this 80% considered to be ncRNA.

In molecular biology, RNA polymerase, is an enzyme that synthesizes RNA from a DNA template.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.
A cDNA library is a combination of cloned cDNA fragments inserted into a collection of host cells, which constitute some portion of the transcriptome of the organism and are stored as a "library". cDNA is produced from fully transcribed mRNA found in the nucleus and therefore contains only the expressed genes of an organism. Similarly, tissue-specific cDNA libraries can be produced. In eukaryotic cells the mature mRNA is already spliced, hence the cDNA produced lacks introns and can be readily expressed in a bacterial cell. While information in cDNA libraries is a powerful and useful tool since gene products are easily identified, the libraries lack information about enhancers, introns, and other regulatory elements found in a genomic DNA library.

Retrotransposons are a type of genetic component that copy and paste themselves into different genomic locations (transposon) by converting RNA back into DNA through the process reverse transcription using an RNA transposition intermediate.

A primary transcript is the single-stranded ribonucleic acid (RNA) product synthesized by transcription of DNA, and processed to yield various mature RNA products such as mRNAs, tRNAs, and rRNAs. The primary transcripts designated to be mRNAs are modified in preparation for translation. For example, a precursor mRNA (pre-mRNA) is a type of primary transcript that becomes a messenger RNA (mRNA) after processing.
Rapid amplification of cDNA ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE results in the production of a cDNA copy of the RNA sequence of interest, produced through reverse transcription, followed by PCR amplification of the cDNA copies. The amplified cDNA copies are then sequenced and, if long enough, should map to a unique genomic region. RACE is commonly followed up by cloning before sequencing of what was originally individual RNA molecules. A more high-throughput alternative which is useful for identification of novel transcript structures, is to sequence the RACE-products by next generation sequencing technologies.
This is a list of topics in molecular biology. See also index of biochemistry articles.
Mung bean nuclease is a nuclease derived from sprouts of the mung bean that removes nucleotides in a step-wise manner from single-stranded DNA molecules (ssDNA) and is used in biotechnological applications to remove such ssDNA from a mixture also containing double-stranded DNA (dsDNA). This enzyme is useful for transcript mapping, removal of single-stranded regions in DNA hybrids or single-stranded overhangs produced by restriction enzymes, etc. It has an activity similar to Nuclease S1, but it has higher specificity for single-stranded molecules.
Nuclease protection assay is a laboratory technique used in biochemistry and genetics to identify individual RNA molecules in a heterogeneous RNA sample extracted from cells. The technique can identify one or more RNA molecules of known sequence even at low total concentration. The extracted RNA is first mixed with antisense RNA or DNA probes that are complementary to the sequence or sequences of interest and the complementary strands are hybridized to form double-stranded RNA. The mixture is then exposed to ribonucleases that specifically cleave only single-stranded RNA but have no activity against double-stranded RNA. When the reaction runs to completion, susceptible RNA regions are degraded to very short oligomers or to individual nucleotides; the surviving RNA fragments are those that were complementary to the added antisense strand and thus contained the sequence of interest.
This glossary of genetics is a list of definitions of terms and concepts commonly used in the study of genetics and related disciplines in biology, including molecular biology and evolutionary biology. It is intended as introductory material for novices; for more specific and technical detail, see the article corresponding to each term. For related terms, see Glossary of evolutionary biology.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. A SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Experimental approaches of determining the structure of nucleic acids, such as RNA and DNA, can be largely classified into biophysical and biochemical methods. Biophysical methods use the fundamental physical properties of molecules for structure determination, including X-ray crystallography, NMR and cryo-EM. Biochemical methods exploit the chemical properties of nucleic acids using specific reagents and conditions to assay the structure of nucleic acids. Such methods may involve chemical probing with specific reagents, or rely on native or analogue chemistry. Different experimental approaches have unique merits and are suitable for different experimental purposes.
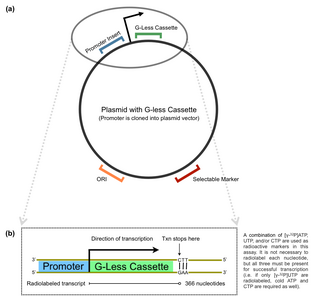
The G-less cassette transcription assay is a method used in molecular biology to determine promoter strength in vitro. The technique involves quantification of an mRNA product with the use of a plasmid. The G-less cassette is part of a pre-constructed vector, usually containing a multiple cloning site (MCS) upstream of the cassette. For this reason, promoters of interest can be inserted directly into the MCS to ultimately measure the accuracy and efficiency of a promoter in recruiting transcription machinery.

RNase H-dependent PCR (rhPCR) is a modification of the standard PCR technique. In rhPCR, the primers are designed with a removable amplification block on the 3’ end. Amplification of the blocked primer is dependent on the cleavage activity of a hyperthermophilic archaeal Type II RNase H enzyme during hybridization to the complementary target sequence. This RNase H enzyme possesses several useful characteristics that enhance the PCR. First, it has very little enzymatic activity at low temperature, enabling a “hot start PCR” without modifications to the DNA polymerase. Second, the cleavage efficiency of the enzyme is reduced in the presence of mismatches near the RNA residue. This allows for reduced primer dimer formation, detection of alternative splicing variants, ability to perform multiplex PCR with higher numbers of PCR primers, and the ability to detect single-nucleotide polymorphisms.
Prime editing is a ‘search-and-replace’ genome editing technology in molecular biology by which the genome of living organisms may be modified. The technology directly writes new genetic information into a targeted DNA site. It uses a fusion protein, consisting of a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase enzyme, and a prime editing guide RNA (pegRNA), capable of identifying the target site and providing the new genetic information to replace the target DNA nucleotides. It mediates targeted insertions, deletions, and base-to-base conversions without the need for double strand breaks (DSBs) or donor DNA templates.
![Overview of the primer extension. Transcript of interest has uracil at +1 (unknown before assay). 2) Synthesize an 5' end-labeled primer (using [g32P] ATP. 3) Make cDNA by extending primer with reverse transcriptase to 5'-end of transcript. 4) Result is a radioactively labeled extended primer cDNA that is denatured and separated on a polyacrylamide gel and detected by autoradiography. Typically, a sequencing ladder using the same primer is also on the gel, allowing for rapid identification of the +1 nucleotide (shown in purple.) Primer Extension Assay.jpg](http://upload.wikimedia.org/wikipedia/commons/thumb/e/eb/Primer_Extension_Assay.jpg/330px-Primer_Extension_Assay.jpg)