
Crystallography is the experimental science of determining the arrangement of atoms in crystalline solids. Crystallography is a fundamental subject in the fields of materials science and solid-state physics. The word crystallography is derived from the Ancient Greek word κρύσταλλος, with its meaning extending to all solids with some degree of transparency, and γράφειν. In July 2012, the United Nations recognised the importance of the science of crystallography by proclaiming that 2014 would be the International Year of Crystallography.

Structural biology is a field that is many centuries old which, as defined by the Journal of Structural Biology, deals with structural analysis of living material at every level of organization. Early structural biologists throughout the 19th and early 20th centuries were primarily only able to study structures to the limit of the naked eye's visual acuity and through magnifying glasses and light microscopes.

X-ray crystallography is the experimental science determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract into many specific directions. By measuring the angles and intensities of these diffracted beams, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal. From this electron density, the mean positions of the atoms in the crystal can be determined, as well as their chemical bonds, their crystallographic disorder, and various other information.
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations. The PDB is overseen by an organization called the Worldwide Protein Data Bank, wwPDB.

A chemical structure of a molecule is a spatial arrangement of its atoms and their chemical bonds. Its determination includes a chemist's specifying the molecular geometry and, when feasible and necessary, the electronic structure of the target molecule or other solid. Molecular geometry refers to the spatial arrangement of atoms in a molecule and the chemical bonds that hold the atoms together and can be represented using structural formulae and by molecular models; complete electronic structure descriptions include specifying the occupation of a molecule's molecular orbitals. Structure determination can be applied to a range of targets from very simple molecules to very complex ones.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
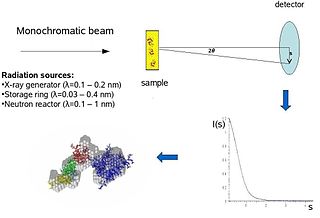
Biological small-angle scattering is a small-angle scattering method for structure analysis of biological materials. Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers. Small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) are the two complementary techniques known jointly as small-angle scattering (SAS). SAS is an analogous method to X-ray and neutron diffraction, wide angle X-ray scattering, as well as to static light scattering. In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles. When used to study biological materials, which are very often in aqueous solution, the scattering pattern is orientation averaged.
In physics, the phase problem is the problem of loss of information concerning the phase that can occur when making a physical measurement. The name comes from the field of X-ray crystallography, where the phase problem has to be solved for the determination of a structure from diffraction data. The phase problem is also met in the fields of imaging and signal processing. Various approaches of phase retrieval have been developed over the years.
Electron crystallography is a method to determine the arrangement of atoms in solids using a transmission electron microscope (TEM). It can involve the use of high-resolution transmission electron microscopy images, electron diffraction patterns including convergent-beam electron diffraction or combinations of these. It has been successful in determining some bulk structures, and also surface structures. Two related methods are low-energy electron diffraction which has solved the structure of many surfaces, and reflection high-energy electron diffraction which is used to monitor surfaces often during growth.

Transmission electron cryomicroscopy (CryoTEM), commonly known as cryo-EM, is a form of cryogenic electron microscopy, more specifically a type of transmission electron microscopy (TEM) where the sample is studied at cryogenic temperatures. Cryo-EM, specifically 3-dimensional electron microscopy (3DEM), is gaining popularity in structural biology.
In crystallography, direct methods are a family of methods for estimating the phases of the Fourier transform of the scattering density from the corresponding magnitudes. The methods generally exploit constraints or statistical correlations between the phases of different Fourier components that result from the fact that the scattering density must be a positive real number.
In structural biology, as well as in virtually all sciences that produce three-dimensional data, the Fourier shell correlation (FSC) measures the normalised cross-correlation coefficient between two 3-dimensional volumes over corresponding shells in Fourier space (i.e., as a function of spatial frequency). The FSC is the three-dimensional extension of the two-dimensional Fourier ring correlation (FRC); also known as: spatial frequency correlation function.
In scientific imaging, the two-dimensional spectral signal-to-noise ratio (SSNR) is a signal-to-noise ratio measure which measures the normalised cross-correlation coefficient between several two-dimensional images over corresponding rings in Fourier space as a function of spatial frequency. It is a multi-particle extension of the Fourier ring correlation (FRC), which is related to the Fourier shell correlation. The SSNR is a popular method for finding the resolution of a class average in cryo-electron microscopy.

Crystallographic image processing (CIP) is traditionally understood as being a set of key steps in the determination of the atomic structure of crystalline matter from high-resolution electron microscopy (HREM) images obtained in a transmission electron microscope (TEM) that is run in the parallel illumination mode. The term was created in the research group of Sven Hovmöller at Stockholm University during the early 1980s and became rapidly a label for the "3D crystal structure from 2D transmission/projection images" approach. Since the late 1990s, analogous and complementary image processing techniques that are directed towards the achieving of goals with are either complementary or entirely beyond the scope of the original inception of CIP have been developed independently by members of the computational symmetry/geometry, scanning transmission electron microscopy, scanning probe microscopy communities, and applied crystallography communities.
The term macromolecular assembly (MA) refers to massive chemical structures such as viruses and non-biologic nanoparticles, cellular organelles and membranes and ribosomes, etc. that are complex mixtures of polypeptide, polynucleotide, polysaccharide or other polymeric macromolecules. They are generally of more than one of these types, and the mixtures are defined spatially, and with regard to their underlying chemical composition and structure. Macromolecules are found in living and nonliving things, and are composed of many hundreds or thousands of atoms held together by covalent bonds; they are often characterized by repeating units. Assemblies of these can likewise be biologic or non-biologic, though the MA term is more commonly applied in biology, and the term supramolecular assembly is more often applied in non-biologic contexts. MAs of macromolecules are held in their defined forms by non-covalent intermolecular interactions, and can be in either non-repeating structures, or in repeating linear, circular, spiral, or other patterns. The process by which MAs are formed has been termed molecular self-assembly, a term especially applied in non-biologic contexts. A wide variety of physical/biophysical, chemical/biochemical, and computational methods exist for the study of MA; given the scale of MAs, efforts to elaborate their composition and structure and discern mechanisms underlying their functions are at the forefront of modern structure science.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
Resolution by Proxy (ResProx) is a method for assessing the equivalent X-ray resolution of NMR-derived protein structures. ResProx calculates resolution from coordinate data rather than from electron density or other experimental inputs. This makes it possible to calculate the resolution of a structure regardless of how it was solved. ResProx was originally designed to serve as a simple, single-number evaluation that allows straightforward comparison between the quality/resolution of X-ray structures and the quality of a given NMR structure. However, it can also be used to assess the reliability of an experimentally reported X-ray structure resolution, to evaluate protein structures solved by unconventional or hybrid means and to identify fraudulent structures deposited in the PDB. ResProx incorporates more than 25 different structural features to determine a single resolution-like value. ResProx values are reported in Angstroms. Tests on thousands of X-ray structures show that ResProx values match very closely to resolution values reported by X-ray crystallographers. Resolution-by-proxy values can be calculated for newly determined protein structures using a freely accessible ResProx web server. This server accepts protein coordinate data and generates a resolution estimate for that input structure.

Cryogenic electron microscopy (cryo-EM) is a cryomicroscopy technique applied on samples cooled to cryogenic temperatures. For biological specimens, the structure is preserved by embedding in an environment of vitreous ice. An aqueous sample solution is applied to a grid-mesh and plunge-frozen in liquid ethane or a mixture of liquid ethane and propane. While development of the technique began in the 1970s, recent advances in detector technology and software algorithms have allowed for the determination of biomolecular structures at near-atomic resolution. This has attracted wide attention to the approach as an alternative to X-ray crystallography or NMR spectroscopy for macromolecular structure determination without the need for crystallization.
Microcrystal electron diffraction, or MicroED, is a CryoEM method that was developed by the Gonen laboratory in late 2013 at the Janelia Research Campus of the Howard Hughes Medical Institute. MicroED is a form of electron crystallography where thin 3D crystals are used for structure determination by electron diffraction. Prior to this demonstration, macromolecular (protein) electron crystallography was only used on 2D crystals, for example.
This is a timeline of crystallography.
