Combinatorial chemistry comprises chemical synthetic methods that make it possible to prepare a large number of compounds in a single process. These compound libraries can be made as mixtures, sets of individual compounds or chemical structures generated by computer software. Combinatorial chemistry can be used for the synthesis of small molecules and for peptides.
Gene silencing is the regulation of gene expression in a cell to prevent the expression of a certain gene. Gene silencing can occur during either transcription or translation and is often used in research. In particular, methods used to silence genes are being increasingly used to produce therapeutics to combat cancer and other diseases, such as infectious diseases and neurodegenerative disorders.
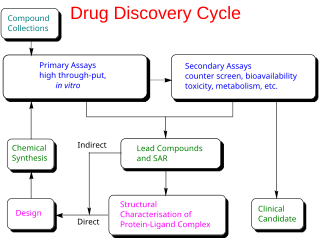
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
grid.org was a website and online community established in 2001 for cluster computing and grid computing software users. For six years it operated several different volunteer computing projects that allowed members to donate their spare computer cycles to worthwhile causes. In 2007, it became a community for open source cluster and grid computing software. After around 2010 it redirected to other sites.

High-throughput screening (HTS) is a method for scientific experimentation especially used in drug discovery and relevant to the fields of biology, materials science and chemistry. Using robotics, data processing/control software, liquid handling devices, and sensitive detectors, high-throughput screening allows a researcher to quickly conduct millions of chemical, genetic, or pharmacological tests. Through this process one can quickly recognize active compounds, antibodies, or genes that modulate a particular biomolecular pathway. The results of these experiments provide starting points for drug design and for understanding the noninteraction or role of a particular location.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
PubChem is a database of chemical molecules and their activities against biological assays. The system is maintained by the National Center for Biotechnology Information (NCBI), a component of the National Library of Medicine, which is part of the United States National Institutes of Health (NIH). PubChem can be accessed for free through a web user interface. Millions of compound structures and descriptive datasets can be freely downloaded via FTP. PubChem contains multiple substance descriptions and small molecules with fewer than 100 atoms and 1,000 bonds. More than 80 database vendors contribute to the growing PubChem database.
Chemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
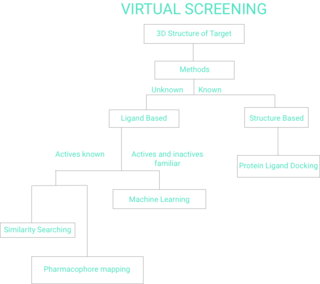
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
High-content screening (HCS), also known as high-content analysis (HCA) or cellomics, is a method that is used in biological research and drug discovery to identify substances such as small molecules, peptides, or RNAi that alter the phenotype of a cell in a desired manner. Hence high content screening is a type of phenotypic screen conducted in cells involving the analysis of whole cells or components of cells with simultaneous readout of several parameters. HCS is related to high-throughput screening (HTS), in which thousands of compounds are tested in parallel for their activity in one or more biological assays, but involves assays of more complex cellular phenotypes as outputs. Phenotypic changes may include increases or decreases in the production of cellular products such as proteins and/or changes in the morphology of the cell. Hence HCA typically involves automated microscopy and image analysis. Unlike high-content analysis, high-content screening implies a level of throughput which is why the term "screening" differentiates HCS from HCA, which may be high in content but low in throughput.
Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD) is a method used for finding lead compounds as part of the drug discovery process. Fragments are small organic molecules which are small in size and low in molecular weight. It is based on identifying small chemical fragments, which may bind only weakly to the biological target, and then growing them or combining them to produce a lead with a higher affinity. FBLD can be compared with high-throughput screening (HTS). In HTS, libraries with up to millions of compounds, with molecular weights of around 500 Da, are screened, and nanomolar binding affinities are sought. In contrast, in the early phase of FBLD, libraries with a few thousand compounds with molecular weights of around 200 Da may be screened, and millimolar affinities can be considered useful. FBLD is a technique being used in research for discovering novel potent inhibitors. This methodology could help to design multitarget drugs for multiple diseases. The multitarget inhibitor approach is based on designing an inhibitor for the multiple targets. This type of drug design opens up new polypharmacological avenues for discovering innovative and effective therapies. Neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s, among others, also show rather complex etiopathologies. Multitarget inhibitors are more appropriate for addressing the complexity of AD and may provide new drugs for controlling the multifactorial nature of AD, stopping its progression.
DNA-encoded chemical libraries (DECL) is a technology for the synthesis and screening on an unprecedented scale of collections of small molecule compounds. DECL is used in medicinal chemistry to bridge the fields of combinatorial chemistry and molecular biology. The aim of DECL technology is to accelerate the drug discovery process and in particular early phase discovery activities such as target validation and hit identification.
Computational Resources for Drug Discovery (CRDD) is one of the important silico modules of Open Source for Drug Discovery (OSDD). The CRDD web portal provides computer resources related to drug discovery on a single platform. It provides computational resources for researchers in computer-aided drug design, a discussion forum, and resources to maintain Wikipedia related to drug discovery, predict inhibitors, and predict the ADME-Tox property of molecules One of the major objectives of CRDD is to promote open source software in the field of chemoinformatics and pharmacoinformatics.

In the field of drug discovery, reverse pharmacology also known as target-based drug discovery (TDD), a hypothesis is first made that modulation of the activity of a specific protein target thought to be disease modifying will have beneficial therapeutic effects. Screening of chemical libraries of small molecules is then used to identify compounds that bind with high affinity to the target. The hits from these screens are then used as starting points for drug discovery. This method became popular after the sequencing of the human genome which allowed rapid cloning and synthesis of large quantities of purified proteins. This method is the most widely used in drug discovery today. Differently than the classical (forward) pharmacology, with the reverse pharmacology approach in vivo efficacy of identified active (lead) compounds is usually performed in the final drug discovery stages.
Phenotypic screening is a type of screening used in biological research and drug discovery to identify substances such as small molecules, peptides, or RNAi that alter the phenotype of a cell or an organism in a desired manner. Phenotypic screening must be followed up with identification and validation, often through the use of chemoproteomics, to identify the mechanisms through which a phenotypic hit works.
In the field of drug discovery, classical pharmacology, also known as forward pharmacology, or phenotypic drug discovery (PDD), relies on phenotypic screening of chemical libraries of synthetic small molecules, natural products or extracts to identify substances that have a desirable therapeutic effect. Using the techniques of medicinal chemistry, the potency, selectivity, and other properties of these screening hits are optimized to produce candidate drugs.
Topological inhibitors are rigid three-dimensional molecules of inorganic, organic, and hybrid compounds that form multicentered supramolecular interactions in vacant cavities of protein macromolecules and their complexes . Extensive surface and very diverse geometry make cage compounds with an encapsulated metal ion (clathrochelates) suitable for targeting both the active and allosteric sites of enzymes as well as the interfaces of their macromolecular complexes. An efficient structure- and concentration-dependent transcription inhibition in a model in vitro systems based on RNA and DNA polymerases by the iron(II) mono- and bis-clathrochelates at their submicro- and nanomolar concentrations, respectively, is observed in. Molecular docking and preincubation experiments suggested that these cage compounds form supramolecular assemblies with protein residues as well as with DNA and RNA. Thus, they are prospective precursors for the design of antiviral and anticancer drug candidates.
Early twenty-first century pesticide research has focused on developing molecules that combine low use rates and that are more selective, safer, resistance-breaking and cost-effective. Obstacles include increasing pesticide resistance and an increasingly stringent regulatory environment.
Chemoproteomics entails a broad array of techniques used to identify and interrogate protein-small molecule interactions. Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery, in which lead compounds are designed to interact with predetermined disease-driving biological targets. As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action. Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.