
A carboxylic acid is an organic acid that contains a carboxyl group (C(=O)OH) attached to an R-group. The general formula of a carboxylic acid is R–COOH or R-CO2H, with R referring to the alkyl, alkenyl, aryl, or other group. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
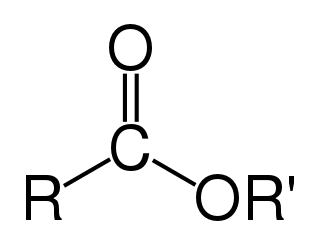
An ester is a chemical compound derived from an acid in which at least one –OH hydroxyl group is replaced by an –O– alkyl (alkoxy) group, as in the substitution reaction of a carboxylic acid and an alcohol. Glycerides are fatty acid esters of glycerol; they are important in biology, being one of the main classes of lipids and comprising the bulk of animal fats and vegetable oils.
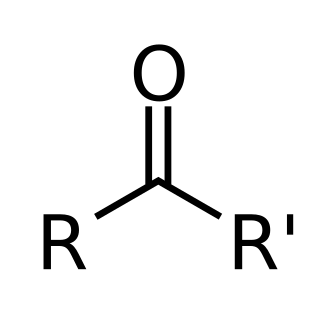
In chemistry, a ketone is a functional group with the structure R2C=O, where R can be a variety of carbon-containing substituents. Ketones contain a carbonyl group (a carbon-oxygen double bond). The simplest ketone is acetone (R = R' = methyl), with the formula CH3C(O)CH3. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids (e.g., testosterone), and the solvent acetone.

The Beckmann rearrangement, named after the German chemist Ernst Otto Beckmann (1853–1923), is a rearrangement of an oxime functional group to substituted amides. The rearrangement has also been successful performed on haloimines and nitrones. Cyclic oximes and haloimines yield lactams.
The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.
Ozonolysis is an organic reaction where the unsaturated bonds of alkenes, alkynes, or azo compounds are cleaved with ozone. Alkenes and alkynes form organic compounds in which the multiple carbon–carbon bond has been replaced by a carbonyl group while azo compounds form nitrosamines. The outcome of the reaction depends on the type of multiple bond being oxidized and the work-up conditions.
A 1,2-rearrangement or 1,2-migration or 1,2-shift or Whitmore 1,2-shift is an organic reaction where a substituent moves from one atom to another atom in a chemical compound. In a 1,2 shift the movement involves two adjacent atoms but moves over larger distances are possible. In the example below the substituent R moves from carbon atom C2 to C3.

Organoborane or organoboron compounds are chemical compounds of boron and carbon that are organic derivatives of BH3, for example trialkyl boranes. Organoboron chemistry or organoborane chemistry is the chemistry of these compounds.

The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.
Nucleophilic acyl substitution describe a class of substitution reactions involving nucleophiles and acyl compounds. In this type of reaction, a nucleophile – such as an alcohol, amine, or enolate – displaces the leaving group of an acyl derivative – such as an acid halide, anhydride, or ester. The resulting product is a carbonyl-containing compound in which the nucleophile has taken the place of the leaving group present in the original acyl derivative. Because acyl derivatives react with a wide variety of nucleophiles, and because the product can depend on the particular type of acyl derivative and nucleophile involved, nucleophilic acyl substitution reactions can be used to synthesize a variety of different products.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994 was the first total synthesis of Taxol.

The Dakin oxidation is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde or ketone reacts with hydrogen peroxide in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidized, and the hydrogen peroxide is reduced.
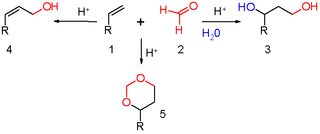
The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol. When water is absent, the cationic intermediate loses a proton to give an allylic alcohol. With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane. When water is replaced by acetic acid the corresponding esters are formed.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The semipinacol rearrangement is a rearrangement reaction in organic chemistry involving a heterosubstituted alcohol of the type R1R2(HO)C–C(X)R3R4. The hetero substituent can be a halogen (Cl, Br, I), a tosylate, a mesylate or a thiol group. This reaction proceeds by removal of the leaving group X forming a carbocation as electron deficient center. One of the adjacent alkyl groups then migrates to the positive carbon in a 1,2-shift. Simultaneously with the shift, a pi bond forms from the oxygen to carbon, assisting in driving the migrating group off its position. The result is a ketone or aldehyde. In another definition all semipinacol rearrangements "share a common reactive species in which an electrophilic carbon center, including but not limited to carbocations, is vicinal to an oxygen-containing carbon and can drive the 1,2-migration of a C–C or C–H bond to terminate the process, generating a carbonyl group ".
The Kornblum–DeLaMare rearrangement is a rearrangement reaction in organic chemistry in which a primary or secondary organic peroxide is converted to the corresponding ketone and alcohol under acid or base catalysis. The reaction is relevant as a tool in organic synthesis and is a key step in the biosynthesis of prostaglandins.
The Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, in organic chemistry is a special type of elimination reaction in which two vicinal substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents unlike regular eliminations which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.

The Jones oxidation is an organic reaction for the oxidation of primary and secondary alcohols to carboxylic acids and ketones, respectively. It is named after its discoverer, Sir Ewart Jones. The reaction was an early method for the oxidation of alcohols. Its use has subsided because milder, more selective reagents have been developed, e.g. Collins reagent.
The Buchner–Curtius–Schlotterbeck reaction is the reaction of aldehydes or ketones with aliphatic diazoalkanes to form homologated ketones. It was first described by Eduard Buchner and Theodor Curtius in 1885 and later by Fritz Schlotterbeck in 1907. Two German chemists also preceded Schlotterbeck in discovery of the reaction, Hans von Pechmann in 1895 and Viktor Meyer in 1905. The reaction has since been extended to the synthesis of β-keto esters from the condensation between aldehydes and diazo esters. The general reaction scheme is as follows:

Trifluoroperacetic acid is an organofluorine compound, the peroxy acid analog of trifluoroacetic acid, with the condensed structural formula CF
3COOOH. It is a strong oxidizing agent for organic oxidation reactions, such as in Baeyer–Villiger oxidations of ketones. It is the most reactive of the organic peroxy acids, allowing it to successfully oxidise relatively unreactive alkenes to epoxides where other peroxy acids are ineffective. It can also oxidise the chalcogens in some functional groups, such as by transforming selenoethers to selones. It is a potentially explosive material and is not commercially available, but it can be quickly prepared as needed. Its use as a laboratory reagent was pioneered and developed by William D. Emmons.
