Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electrophilic double or triple bond reacts with a nucleophile, such that the double or triple bond is broken. Nucleophilic additions differ from electrophilic additions in that the former reactions involve the group to which atoms are added accepting electron pairs, whereas the latter reactions involve the group donating electron pairs.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.
The Shapiro reaction or tosylhydrazone decomposition is an organic reaction in which a ketone or aldehyde is converted to an alkene through an intermediate hydrazone in the presence of 2 equivalents of organolithium reagent. The reaction was discovered by Robert H. Shapiro in 1967. The Shapiro reaction was used in the Nicolaou Taxol total synthesis. This reaction is very similar to the Bamford–Stevens reaction, which also involves the basic decomposition of tosyl hydrazones.

The Peterson olefination is the chemical reaction of α-silyl carbanions with ketones to form a β-hydroxysilane (2) which eliminates to form alkenes (3).

The Seyferth–Gilbert homologation is a chemical reaction of an aryl ketone 1 with dimethyl (diazomethyl)phosphonate 2 and potassium tert-butoxide to give substituted alkynes 3. Dimethyl (diazomethyl)phosphonate 2 is often called the Seyferth–Gilbert reagent.

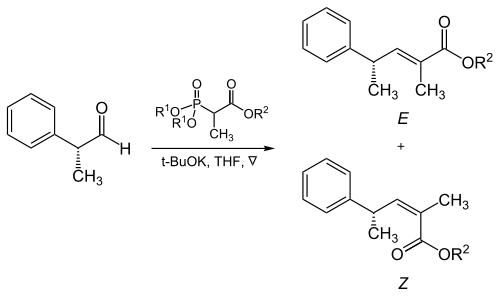
Asymmetric induction describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment. Asymmetric induction is a key element in asymmetric synthesis.
The Julia olefination (also known as the Julia–Lythgoe olefination) is the chemical reaction used in organic chemistry of phenyl sulfones (1) with aldehydes (or ketones) to give alkenes (olefins)(3) after alcohol functionalization and reductive elimination using sodium amalgam or SmI2. The reaction is named after the French chemist Marc Julia.
The Meyer–Schuster rearrangement is the chemical reaction described as an acid-catalyzed rearrangement of secondary and tertiary propargyl alcohols to α,β-unsaturated ketones if the alkyne group is internal and α,β-unsaturated aldehydes if the alkyne group is terminal. Reviews have been published by Swaminathan and Narayan, Vartanyan and Banbanyan, and Engel and Dudley, the last of which describes ways to promote the Meyer–Schuster rearrangement over other reactions available to propargyl alcohols.
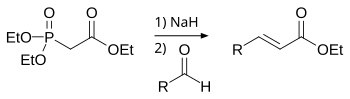
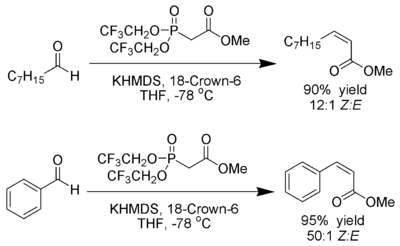
William D. Emmons was an American chemist and published with William S. Wadsworth a modification to the Wittig-Horner reaction using phosphonate-stabilized carbanions, now called the Horner-Wadsworth-Emmons reaction or HWE reaction or Horner-Wittig reaction.
The Abramov reaction is the related conversions of trialkyl to α-hydroxy phosphonates by the addition to carbonyl compounds. In terms of mechanism, the reaction involves attack of the nucleophilic phosphorus atom on the carbonyl carbon. It was named after the Russian chemist Vasilii Semenovich Abramov (1904–1968) in 1957.
Electrophilic substitution of unsaturated silanes involves attack of an electrophile on an allyl- or vinylsilane. An allyl or vinyl group is incorporated at the electrophilic center after loss of the silyl group.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
In organic chemistry, Wittig reagents are organophosphorus compounds of the formula R3P=CHR', where R is usually phenyl. They are used to convert ketones and aldehydes to alkenes: