Phylogenetic footprinting is a technique used to identify transcription factor binding sites (TFBS) within a non-coding region of DNA of interest by comparing it to the orthologous sequence in different species. When this technique is used with a large number of closely related species, this is called phylogenetic shadowing. [1]

In molecular biology, a transcription factor (TF) is a protein that controls the rate of transcription of genetic information from DNA to messenger RNA, by binding to a specific DNA sequence. The function of TFs is to regulate—turn on and off—genes in order to make sure that they are expressed in the right cell at the right time and in the right amount throughout the life of the cell and the organism. Groups of TFs function in a coordinated fashion to direct cell division, cell growth, and cell death throughout life; cell migration and organization during embryonic development; and intermittently in response to signals from outside the cell, such as a hormone. There are up to 2600 TFs in the human genome.

In mathematics, a sequence is an enumerated collection of objects in which repetitions are allowed. Like a set, it contains members. The number of elements is called the length of the sequence. Unlike a set, the same elements can appear multiple times at different positions in a sequence, and order matters. Formally, a sequence can be defined as a function whose domain is either the set of the natural numbers or the set of the first n natural numbers. The position of an element in a sequence is its rank or index; it is the natural number from which the element is the image. It depends on the context or a specific convention, if the first element has index 0 or 1. When a symbol has been chosen for denoting a sequence, the nth element of the sequence is denoted by this symbol with n as subscript; for example, the nth element of the Fibonacci sequence is generally denoted Fn.
In biology, a species ( ) is the basic unit of classification and a taxonomic rank of an organism, as well as a unit of biodiversity. A species is often defined as the largest group of organisms in which any two individuals of the appropriate sexes or mating types can produce fertile offspring, typically by sexual reproduction. Other ways of defining species include their karyotype, DNA sequence, morphology, behaviour or ecological niche. In addition, paleontologists use the concept of the chronospecies since fossil reproduction cannot be examined. While these definitions may seem adequate, when looked at more closely they represent problematic species concepts. For example, the boundaries between closely related species become unclear with hybridisation, in a species complex of hundreds of similar microspecies, and in a ring species. Also, among organisms that reproduce only asexually, the concept of a reproductive species breaks down, and each clone is potentially a microspecies.
Contents
- History
- Protocol
- Not all TFBS are found
- Species specific binding sites
- Very short binding sites
- Less specific binding factors
- Not enough data
- Compound binding regions
- Accuracy
- References
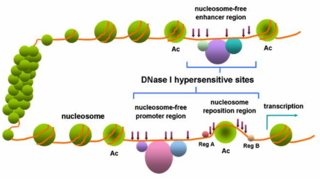
Researchers have found that non-coding pieces of DNA contain binding sites for regulatory proteins that govern the spatiotemporal expression of genes. These transcription factor binding sites (TFBS), or regulatory motifs, have proven hard to identify, primarily because they are short in length, and can show sequence variation. The importance of understanding transcriptional regulation to many fields of biology has led researchers to develop strategies for predicting the presence of TFBS, many of which have led to publicly available databases. One such technique is Phylogenetic Footprinting.

Deoxyribonucleic acid is a molecule composed of two chains that coil around each other to form a double helix carrying the genetic instructions used in the growth, development, functioning, and reproduction of all known organisms and many viruses. DNA and ribonucleic acid (RNA) are nucleic acids; alongside proteins, lipids and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life.
Biology is the natural science that studies life and living organisms, including their physical structure, chemical processes, molecular interactions, physiological mechanisms, development and evolution. Despite the complexity of the science, there are certain unifying concepts that consolidate it into a single, coherent field. Biology recognizes the cell as the basic unit of life, genes as the basic unit of heredity, and evolution as the engine that propels the creation and extinction of species. Living organisms are open systems that survive by transforming energy and decreasing their local entropy to maintain a stable and vital condition defined as homeostasis.
Footprinting is the technique used for gathering information about computer systems and the entities they belong to. To get this information, a hacker might use various tools and technologies. This information is very useful to a hacker who is trying to crack a whole system.
Phylogenetic footprinting relies upon two major concepts:
- The function and DNA binding preferences of transcription factors are well-conserved between diverse species.
- Important non-coding DNA sequences that are essential for regulating gene expression will show differential selective pressure. A slower rate of change occurs in TFBS than in other, less critical, parts of the non-coding genome. [2]