In genetics, complementary DNA (cDNA) is DNA that was reverse transcribed from an RNA. cDNA exists in both single-stranded and double-stranded forms and in both natural and engineered forms.
In genetics and biochemistry, sequencing means to determine the primary structure of an unbranched biopolymer. Sequencing results in a symbolic linear depiction known as a sequence which succinctly summarizes much of the atomic-level structure of the sequenced molecule.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.

DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
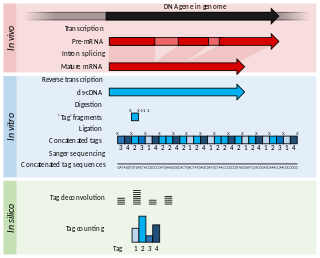
Serial Analysis of Gene Expression (SAGE) is a transcriptomic technique used by molecular biologists to produce a snapshot of the messenger RNA population in a sample of interest in the form of small tags that correspond to fragments of those transcripts. Several variants have been developed since, most notably a more robust version, LongSAGE, RL-SAGE and the most recent SuperSAGE. Many of these have improved the technique with the capture of longer tags, enabling more confident identification of a source gene.

2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.

RNA-Seq is a technique that uses next-generation sequencing to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Cap analysis of gene expression (CAGE) is a gene expression technique used in molecular biology to produce a snapshot of the 5′ end of the messenger RNA population in a biological sample. The small fragments from the very beginnings of mRNAs are extracted, reverse-transcribed to cDNA, PCR amplified and sequenced. CAGE was first published by Hayashizaki, Carninci and co-workers in 2003. CAGE has been extensively used within the FANTOM research projects.
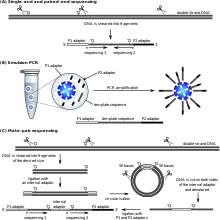
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Magnetic sequencing is a single-molecule sequencing method in development. A DNA hairpin, containing the sequence of interest, is bound between a magnetic bead and a glass surface. A magnetic field is applied to stretch the hairpin open into single strands, and the hairpin refolds after decreasing of the magnetic field. The hairpin length can be determined by direct imaging of the diffraction rings of the magnetic beads using a simple microscope. The DNA sequences are determined by measuring the changes in the hairpin length following successful hybridization of complementary nucleotides.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
G&T-seq is a novel form of single cell sequencing technique allowing one to simultaneously obtain both transcriptomic and genomic data from single cells, allowing for direct comparison of gene expression data to its corresponding genomic data in the same cell...

Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double-stranded DNA to detect mutations with higher accuracy and lower error rates.

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

Spatial transcriptomics is a method for assigning cell types to their locations in the histological sections and can also be used to determine subcellular localization of mRNA molecules. First described in 2016 by Ståhl et al., it has since undergone a variety of improvements and modifications.
CITE-Seq is a method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level. So far, the method has been demonstrated to work with only a few proteins per cell. As such, it provides an additional layer of information for the same cell by combining both proteomics and transcriptomics data. For phenotyping, this method has been shown to be as accurate as flow cytometry by the groups that developed it. It is currently one of the main methods, along with REAP-Seq, to evaluate both gene expression and protein levels simultaneously in different species.