
A covalent bond is a chemical bond that involves the sharing of electrons to form electron pairs between atoms. These electron pairs are known as shared pairs or bonding pairs. The stable balance of attractive and repulsive forces between atoms, when they share electrons, is known as covalent bonding. For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full valence shell, corresponding to a stable electronic configuration. In organic chemistry, covalent bonding is much more common than ionic bonding.

In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.
In organic chemistry, Markovnikov's rule or Markownikoff's rule describes the outcome of some addition reactions. The rule was formulated by Russian chemist Vladimir Markovnikov in 1870.
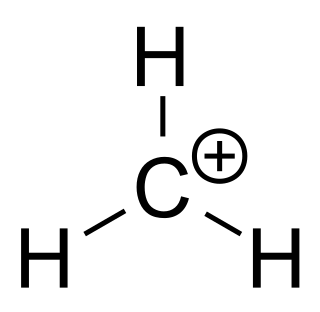
A carbocation is an ion with a positively charged carbon atom. Among the simplest examples are the methenium CH+
3, methanium CH+
5 and vinyl C
2H+
3 cations. Occasionally, carbocations that bear more than one positively charged carbon atom are also encountered.
In chemistry, resonance, also called mesomerism, is a way of describing bonding in certain molecules or polyatomic ions by the combination of several contributing structures into a resonance hybrid in valence bond theory. It has particular value for analyzing delocalized electrons where the bonding cannot be expressed by one single Lewis structure.
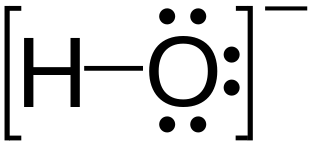
In chemistry, a lone pair refers to a pair of valence electrons that are not shared with another atom in a covalent bond and is sometimes called an unshared pair or non-bonding pair. Lone pairs are found in the outermost electron shell of atoms. They can be identified by using a Lewis structure. Electron pairs are therefore considered lone pairs if two electrons are paired but are not used in chemical bonding. Thus, the number of electrons in lone pairs plus the number of electrons in bonds equals the number of valence electrons around an atom.
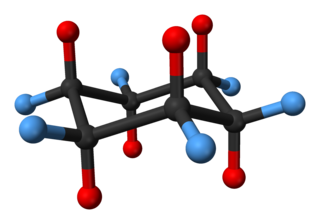
Cyclohexane conformations are any of several three-dimensional shapes adopted by molecules of cyclohexane. Because many compounds feature structurally similar six-membered rings, the structure and dynamics of cyclohexane are important prototypes of a wide range of compounds.

In chemistry, conformational isomerism is a form of stereoisomerism in which the isomers can be interconverted just by rotations about formally single bonds. While any two arrangements of atoms in a molecule that differ by rotation about single bonds can be referred to as different conformations, conformations that correspond to local minima on the potential energy surface are specifically called conformational isomers or conformers. Conformations that correspond to local maxima on the energy surface are the transition states between the local-minimum conformational isomers. Rotations about single bonds involve overcoming a rotational energy barrier to interconvert one conformer to another. If the energy barrier is low, there is free rotation and a sample of the compound exists as a rapidly equilibrating mixture of multiple conformers; if the energy barrier is high enough then there is restricted rotation, a molecule may exist for a relatively long time period as a stable rotational isomer or rotamer. When the time scale for interconversion is long enough for isolation of individual rotamers, the isomers are termed atropisomers. The ring-flip of substituted cyclohexanes constitutes another common form of conformational isomerism.

In chemistry an eclipsed conformation is a conformation in which two substituents X and Y on adjacent atoms A, B are in closest proximity, implying that the torsion angle X–A–B–Y is 0°. Such a conformation can exist in any open chain, single chemical bond connecting two sp3-hybridised atoms, and it is normally a conformational energy maximum. This maximum is often explained by steric hindrance, but its origins sometimes actually lie in hyperconjugation.

In organic chemistry, hyperconjugation refers to the delocalization of electrons with the participation of bonds of primarily σ-character. Usually, hyperconjugation involves the interaction of the electrons in a sigma (σ) orbital with an adjacent unpopulated non-bonding p or antibonding σ* or π* orbitals to give a pair of extended molecular orbitals. However, sometimes, low-lying antibonding σ* orbitals may also interact with filled orbitals of lone pair character (n) in what is termed negative hyperconjugation. Increased electron delocalization associated with hyperconjugation increases the stability of the system. In particular, the new orbital with bonding character is stabilized, resulting in an overall stabilization of the molecule. Only electrons in bonds that are in the β position can have this sort of direct stabilizing effect — donating from a sigma bond on an atom to an orbital in another atom directly attached to it. However, extended versions of hyperconjugation can be important as well. The Baker–Nathan effect, sometimes used synonymously for hyperconjugation, is a specific application of it to certain chemical reactions or types of structures.
In organic chemistry, neighbouring group participation has been defined by the International Union of Pure and Applied Chemistry (IUPAC) as the interaction of a reaction centre with a lone pair of electrons in an atom or the electrons present in a pi bond contained within the parent molecule but not conjugated with the reaction centre. When NGP is in operation it is normal for the reaction rate to be increased. It is also possible for the stereochemistry of the reaction to be abnormal when compared with a normal reaction. While it is possible for neighbouring groups to influence many reactions in organic chemistry this page is limited to neighbouring group effects seen with carbocations and SN2 reactions.

In organic chemistry, the anomeric effect or Edward-Lemieux effect is a stereoelectronic effect that describes the tendency of heteroatomic substituents adjacent to a heteroatom within a cyclohexane ring to prefer the axial orientation instead of the less hindered equatorial orientation that would be expected from steric considerations. This effect was originally observed in pyranose rings by J. T. Edward in 1955 when studying carbohydrate chemistry.

In the study of conformational isomerism, the Gauche effect is an atypical situation where a gauche conformation is more stable than the anti conformation (180°).
The beta-silicon effect in organosilicon chemistry, also called silicon hyperconjugation, is a special type of hyperconjugation that describes the stabilizing influence of a silicon atom on the development of positive charge at a carbon atom one position removed (β) from the silicon atom. The C-Si σ orbital is said to partially overlap with the σ* anti-bonding orbital of the C-leaving group, lowering the energy of the transition state leading to the formation of a carbocation. A prerequisite for the hyperconjugation to occur is an antiperiplanar relationship between the Si group and the leaving group. This allows for the maximum overlap between the C-Si σ orbital and the σ* anti-bonding orbital of the leaving group. Silicon hyperconjugation explains specific observations regarding chemical kinetics and stereochemistry of organic reactions with reactants containing silicon.
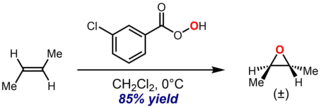
The Prilezhaev reaction, also known as the Prileschajew reaction or Prilezhaev epoxidation, is the chemical reaction of an alkene with a peroxy acid to form epoxides. It is named after Nikolai Prilezhaev, who first reported this reaction in 1909. A widely used peroxy acid for this reaction is meta-chloroperoxybenzoic acid (m-CPBA), due to its stability and good solubility in most organic solvents. The reaction is performed in inert solvents (C6H14, C6H6, CH2Cl2, CHCl3, CCl4) between -10 and 60 °C with the yield of 60-80%.
In chemistry, a radical, also known as a free radical, is an atom, molecule, or ion that has at least one unpaired valence electron. With some exceptions, these unpaired electrons make radicals highly chemically reactive. Many radicals spontaneously dimerize. Most organic radicals have short lifetimes.
In organic chemistry, negative hyperconjugation is the donation of electron density from a filled π- or p-orbital to a neighboring σ*-orbital. This phenomenon, a type of resonance, can stabilize the molecule or transition state. It also causes an elongation of the σ-bond by adding electron density to its antibonding orbital.
In stereochemistry, the Klyne–Prelog system for describing conformations about a single bond offers a more systematic means to unambiguously name complex structures, where the torsional or dihedral angles are not found to occur in 60° increments. Klyne notation views the placement of the substituent on the front atom as being in regions of space called anti/syn and clinal/periplanar relative to a reference group on the rear atom. A plus (+) or minus (–) sign is placed at the front to indicate the sign of the dihedral angle. Anti or syn indicates the substituents are on opposite sides or the same side, respectively. Clinal substituents are found within 30° of either side of a dihedral angle of 60°, 120° (90°–150°), 240° (210°–270°), or 300° (270°–330°). Periplanar substituents are found within 30° of either 0° (330°–30°) or 180° (150°–210°). Juxtaposing the designations produces the following terms for the conformers of butane : gauche butane is syn-clinal, anti butane is anti-periplanar, and eclipsed butane is syn-periplanar.
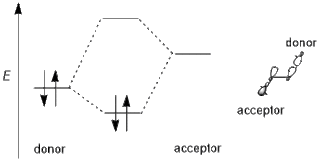
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
In organic chemistry, the Cieplak effect is a predictive model to rationalize why nucleophiles preferentially add to one face of a carbonyl over another. Proposed by Andrzej Stanislaw Cieplak in 1980, it correctly predicts results that could not be justified by the other standard models at the time, such as the Cram and Felkin–Anh models. In the Cieplak model, electrons from a neighboring bond delocalize into the forming carbon–nucleophile (C–Nuc) bond, lowering the energy of the transition state and accelerating the rate of reaction. Whichever bond can best donate its electrons into the C–Nuc bond determines which face of the carbonyl the nucleophile will add to. The nucleophile may be any of a number of reagents, most commonly organometallic or reducing agents. The Cieplak effect is subtle, and often competes with sterics, solvent effects, counterion complexation of the carbonyl oxygen, and other effects to determine product distribution. Subsequent work has questioned its legitimacy.











{kind=link}