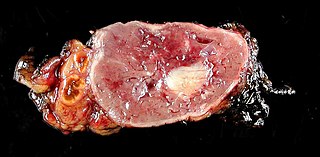
Pheochromocytoma is a rare tumor of the adrenal medulla composed of chromaffin cells, also known as pheochromocytes. When a tumor composed of the same cells as a pheochromocytoma develops outside the adrenal gland, it is referred to as a paraganglioma. These neuroendocrine tumors typically release massive amounts of catecholamines which result in the most common symptoms, including hypertension, tachycardia, and sweating. Rarely, some tumors may secrete little to no catecholamines, making diagnosis difficult. While tumors of the head and neck are parasympathetic, their sympathetic counterparts are predominantly located in the abdomen and pelvis, particularly concentrated at the organ of Zuckerkandl.

Zollinger–Ellison syndrome is rare disease in which tumors cause the stomach to produce too much acid, resulting in peptic ulcers. Symptoms include abdominal pain and diarrhea.
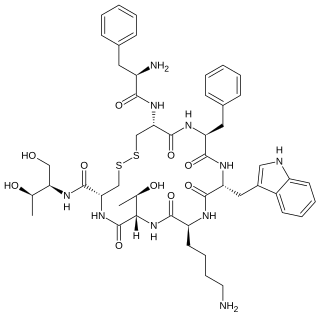
Octreotide, sold under the brand name Sandostatin among others, is an octapeptide that mimics natural somatostatin pharmacologically, though it is a more potent inhibitor of growth hormone, glucagon, and insulin than the natural hormone. It was first synthesized in 1979 by the chemist Wilfried Bauer, and binds predominantly to the somatostatin receptors SSTR2 and SSTR5.

Enterochromaffin (EC) cells are a type of enteroendocrine cell, and neuroendocrine cell. They reside alongside the epithelium lining the lumen of the digestive tract and play a crucial role in gastrointestinal regulation, particularly intestinal motility and secretion. They were discovered by Nikolai Kulchitsky.
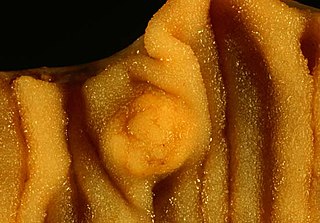
A carcinoid is a slow-growing type of neuroendocrine tumor originating in the cells of the neuroendocrine system. In some cases, metastasis may occur. Carcinoid tumors of the midgut are associated with carcinoid syndrome.

Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. Individuals suffering from this disorder are prone to developing multiple endocrine and nonendocrine tumors. It was first described by Paul Wermer in 1954.
A VIPoma or vipoma is a rare endocrine tumor that overproduces vasoactive intestinal peptide. The incidence is about 1 per 10,000,000 per year. VIPomas usually originate from the non-β islet cells of the pancreas. They are sometimes associated with multiple endocrine neoplasia type 1. Roughly 50–75% of VIPomas are malignant, but even when they are benign, they are problematic because they tend to cause a specific syndrome: the massive amounts of VIP cause a syndrome of profound and chronic watery diarrhea and resultant dehydration, hypokalemia, achlorhydria, acidosis, flushing and hypotension, hypercalcemia, and hyperglycemia. This syndrome is called Verner–Morrison syndrome (VMS), WDHA syndrome, or pancreatic cholera syndrome (PCS). The eponym reflects the physicians who first described the syndrome.

Gastrinomas are neuroendocrine tumors (NETs), usually located in the duodenum or pancreas, that secrete gastrin and cause a clinical syndrome known as Zollinger–Ellison syndrome (ZES). A large number of gastrinomas develop in the pancreas or duodenum, with near-equal frequency, and approximately 10% arise as primary neoplasms in lymph nodes of the pancreaticoduodenal region.

5-Hydroxyindoleacetic acid (5-HIAA) is the main metabolite of serotonin. In chemical analysis of urine samples, 5-HIAA is used to determine serotonin levels in the body.

Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung, and the rest of the body.
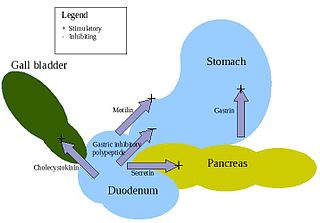
Enteroendocrine cells are specialized cells of the gastrointestinal tract and pancreas with endocrine function. They produce gastrointestinal hormones or peptides in response to various stimuli and release them into the bloodstream for systemic effect, diffuse them as local messengers, or transmit them to the enteric nervous system to activate nervous responses. Enteroendocrine cells of the intestine are the most numerous endocrine cells of the body. They constitute an enteric endocrine system as a subset of the endocrine system just as the enteric nervous system is a subset of the nervous system. In a sense they are known to act as chemoreceptors, initiating digestive actions and detecting harmful substances and initiating protective responses. Enteroendocrine cells are located in the stomach, in the intestine and in the pancreas. Microbiota play key roles in the intestinal immune and metabolic responses in these enteroendocrine cells via their fermentation product, acetate.
Cardiac fibrosis commonly refers to the excess deposition of extracellular matrix in the cardiac muscle, but the term may also refer to an abnormal thickening of the heart valves due to inappropriate proliferation of cardiac fibroblasts. Fibrotic cardiac muscle is stiffer and less compliant and is seen in the progression to heart failure. The description below focuses on a specific mechanism of valvular pathology but there are other causes of valve pathology and fibrosis of the cardiac muscle.
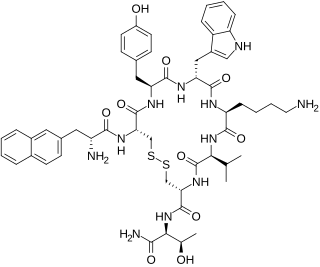
Lanreotide, sold under the brand name Somatuline among others, is a medication used in the management of acromegaly and symptoms caused by neuroendocrine tumors, most notably carcinoid syndrome. It is a long-acting analogue of somatostatin, like octreotide.
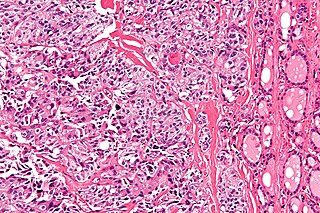
Medullary thyroid cancer is a form of thyroid carcinoma which originates from the parafollicular cells, which produce the hormone calcitonin. Medullary tumors are the third most common of all thyroid cancers and together make up about 3% of all thyroid cancer cases. MTC was first characterized in 1959.

Acromegaly is a disorder that results in excess growth of certain parts of the human body. It is caused by excess growth hormone (GH) after the growth plates have closed. The initial symptom is typically enlargement of the hands and feet. There may also be an enlargement of the forehead, jaw, and nose. Other symptoms may include joint pain, thicker skin, deepening of the voice, headaches, and problems with vision. Complications of the disease may include type 2 diabetes, sleep apnea, and high blood pressure.

Pancreatic neuroendocrine tumours, often referred to as "islet cell tumours", or "pancreatic endocrine tumours" are neuroendocrine neoplasms that arise from cells of the endocrine (hormonal) and nervous system within the pancreas.
Hepatic artery embolization, also known as trans-arterial embolization (TAE), is one of the several therapeutic methods to treat primary liver tumors or metastases to the liver. The embolization therapy can reduce the size of the tumor, and decrease the tumor's impact such its hormone production, effectively decreasing symptoms. The treatment was initially developed in the early 1970s. The several types of hepatic artery treatments are based on the observation that tumor cells get nearly all their nutrients from the hepatic artery, while the normal cells of the liver get about 70-80 percent of their nutrients and 50% their oxygen supply from the portal vein, and thus can survive with the hepatic artery effectively blocked. In practice, hepatic artery embolization occludes the blood flow to the tumors, achieving significant tumor shrinkage in over 80% of people. Shrinkage rates vary.
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is a diffuse parenchymal lung disease which often presents with symptoms of cough and shortness of breath. The pathological definition published by the World Health Organization is “a generalized proliferation of scattered single cells, small nodules, or linear proliferations of pulmonary neuroendocrine (PNE) cells that may be confined to the bronchial and bronchiolar epithelium.” The true prevalence of this disease is not known. To date, just under 200 cases have been reported in the literature. However, with an increase in recognition of this disease by radiologists and pulmonologists, the number of cases has been increasing. DIPNECH predominantly affects middle-aged women with slowly progressive lung obstruction. DIPNECH is usually discovered in one of two ways: 1) as an unexpected finding following a lung surgery; or 2) by evaluation of a patient in a pulmonary clinic with longstanding, unexplained symptoms.
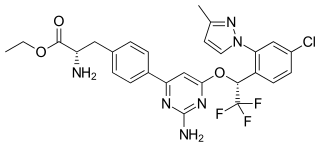
Telotristat ethyl is a prodrug of telotristat, which is an inhibitor of tryptophan hydroxylase. It is formulated as telotristat etiprate — a hippurate salt of telotristat ethyl.
A small intestine neuroendocrine tumor is a carcinoid in the distal small intestine or the proximal large intestine. It is a relatively rare cancer and is diagnosed in approximately 1/100000 people every year. In recent decades the incidence has increased. The prognosis is comparatively good with a median survival of more than 8 years. The disease was named by Siegfried Oberndorfer, a German pathologist, in 1907.



