Related Research Articles

A picture archiving and communication system (PACS) is a medical imaging technology which provides economical storage and convenient access to images from multiple modalities. Electronic images and reports are transmitted digitally via PACS; this eliminates the need to manually file, retrieve, or transport film jackets, the folders used to store and protect X-ray film. The universal format for PACS image storage and transfer is DICOM. Non-image data, such as scanned documents, may be incorporated using consumer industry standard formats like PDF, once encapsulated in DICOM. A PACS consists of four major components: The imaging modalities such as X-ray plain film (PF), computed tomography (CT) and magnetic resonance imaging (MRI), a secured network for the transmission of patient information, workstations for interpreting and reviewing images, and archives for the storage and retrieval of images and reports. Combined with available and emerging web technology, PACS has the ability to deliver timely and efficient access to images, interpretations, and related data. PACS reduces the physical and time barriers associated with traditional film-based image retrieval, distribution, and display.
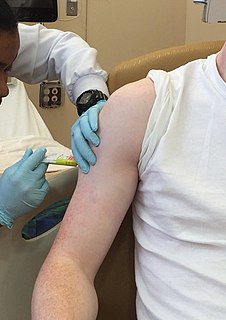
Clinical trials are experiments or observations done in clinical research. Such prospective biomedical or behavioral research studies on human participants are designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.
An electronic health record (EHR) is the systematized collection of patient and population electronically stored health information in a digital format. These records can be shared across different health care settings. Records are shared through network-connected, enterprise-wide information systems or other information networks and exchanges. EHRs may include a range of data, including demographics, medical history, medication and allergies, immunization status, laboratory test results, radiology images, vital signs, personal statistics like age and weight, and billing information.
Clinical audit is a process that has been defined as a quality improvement process that seeks to improve patient care and outcomes through systematic review of care against explicit criteria and the implementation of change
Clinical monitoring is the oversight and administrative efforts that monitor a participant's health and efficacy of the treatment during a clinical trial. Both independent and government-run grant-funding agencies, such as the National Institutes of Health (NIH) and the World Health Organization (WHO), require data and safety monitoring protocols for Phase I and II clinical trials conforming to their standards.
An adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event (AE) can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product.
Acquisition or collection of clinical trial data can be achieved through various methods that may include, but are not limited to, any of the following: paper or electronic medical records, paper forms completed at a site, interactive voice response systems, local electronic data capture systems, or central web based systems.
The Clinical Data Interchange Standards Consortium (CDISC) is a standards developing organization (SDO) dealing with medical research data linked with healthcare, to "enable information system interoperability to improve medical research and related areas of healthcare". The standards support medical research from protocol through analysis and reporting of results and have been shown to decrease resources needed by 60% overall and 70–90% in the start-up stages when they are implemented at the beginning of the research process.
An electronic data capture (EDC) system is a computerized system designed for the collection of clinical data in electronic format for use mainly in human clinical trials. EDC replaces the traditional paper-based data collection methodology to streamline data collection and expedite the time to market for drugs and medical devices. EDC solutions are widely adopted by pharmaceutical companies and contract research organizations (CRO).
A data monitoring committee (DMC) – sometimes called a data and safety monitoring board (DSMB) – is an independent group of experts who monitor patient safety and treatment efficacy data while a clinical trial is ongoing.
A data clarification form (DCF) or data query form is a questionnaire specifically used in clinical research. The DCF is the primary data clarification tool from the trial sponsor or contract research organization (CRO) towards the investigator to clarify discrepancies and ask the investigator for clarification. The DCF is part of the data validation process in a clinical trial.
A clinical data management system or CDMS is a tool used in clinical research to manage the data of a clinical trial. The clinical trial data gathered at the investigator site in the case report form are stored in the CDMS. To reduce the possibility of errors due to human entry, the systems employ various means to verify the data. Systems for clinical data management can be self-contained or part of the functionality of a CTMS. A CTMS with clinical data management functionality can help with the validation of clinical data as well as helps the site employ for other important activities like building patient registries and assist in patient recruitment efforts.
In natural and social science research, a protocol is most commonly a predefined procedural method in the design and implementation of an experiment. Protocols are written whenever it is desirable to standardize a laboratory method to ensure successful replication of results by others in the same laboratory or by other laboratories. Additionally, and by extension, protocols have the advantage of facilitating the assessment of experimental results through peer review. In addition to detailed procedures, equipment, and instruments, protocols will also contain study objectives, reasoning for experimental design, reasoning for chosen sample sizes, safety precautions, and how results were calculated and reported, including statistical analysis and any rules for predefining and documenting excluded data to avoid bias.
A Clinical Research Coordinator (CRC) is a person responsible for conducting clinical trials using good clinical practice (GCP) under the auspices of a Principal Investigator (PI).
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:
Caisis is an open-source, web-based, patient data management system that integrates research with patient care. The system is freely distributed to promote the collection of standard, well structured data suitable for research and multi-institution collaboration.
A clinical trial portal is a web portal or enterprise portal that primarily serves sponsors and investigators in a clinical trial. Clinical portals can be developed for a particular study, however study-specific portals may be part of larger, clinical sponsor or Contract Research Organization (CRO) portals that cover multiple trials. A clinical portal is typically developed by a sponsor or CRO to facilitate centralized access to relevant information, documentation and online applications by investigational sites participating in a trial, as well as for the monitors, study managers, data managers, medical, safety and regulatory staff that help plan, conduct, manage and review the trial.
An electronic patient-reported outcome (ePRO) is a patient-reported outcome that is collected by electronic methods. ePRO methods are most commonly used in clinical trials, but they are also used elsewhere in health care. As a function of the regulatory process, a majority of ePRO questionnaires undergo the linguistic validation process. When the data is captured for a clinical trial, the data is considered a form of Electronic Source Data
Clinical data management (CDM) is a critical process in clinical research, which leads to generation of high-quality, reliable, and statistically sound data from clinical trials. Clinical data management ensures collection, integration and availability of data at appropriate quality and cost. It also supports the conduct, management and analysis of studies across the spectrum of clinical research as defined by the National Institutes of Health (NIH). The ultimate goal of CDM is to ensure that conclusions drawn from research are well supported by the data. Achieving this goal protects public health and confidence in marketed therapeutics.
References
- ↑ "Case Report Form (CRF)". Archived from the original on 2016-01-15.
- Debbie Kennedy, CRF Designer, Canary Publications, ISBN 0-9531174-7-2