A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. When chemical reactions occur, the atoms are rearranged and the reaction is accompanied by an energy change as new products are generated. Classically, chemical reactions encompass changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no change to the nuclei, and can often be described by a chemical equation. Nuclear chemistry is a sub-discipline of chemistry that involves the chemical reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.

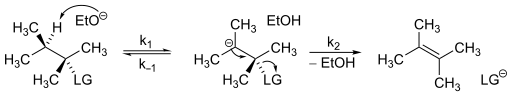
An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.
The unimolecular nucleophilic substitution (SN1) reaction is a substitution reaction in organic chemistry. The Hughes-Ingold symbol of the mechanism expresses two properties—"SN" stands for "nucleophilic substitution", and the "1" says that the rate-determining step is unimolecular. Thus, the rate equation is often shown as having first-order dependence on the substrate and zero-order dependence on the nucleophile. This relationship holds for situations where the amount of nucleophile is much greater than that of the intermediate. Instead, the rate equation may be more accurately described using steady-state kinetics. The reaction involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides under strongly basic conditions or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary and secondary alkyl halides, the alternative SN2 reaction occurs. In inorganic chemistry, the SN1 reaction is often known as the dissociative substitution. This dissociation pathway is well-described by the cis effect. A reaction mechanism was first proposed by Christopher Ingold et al. in 1940. This reaction does not depend much on the strength of the nucleophile, unlike the SN2 mechanism. This type of mechanism involves two steps. The first step is the ionization of alkyl halide in the presence of aqueous acetone or ethyl alcohol. This step provides a carbocation as an intermediate.
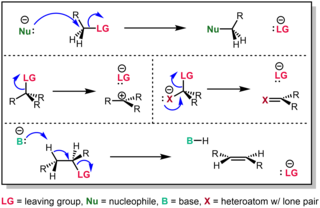
In chemistry, a leaving group is defined by the IUPAC as an atom or group of atoms that detaches from the main or residual part of a substrate during a reaction or elementary step of a reaction. However, in common usage, the term is often limited to a fragment that departs with a pair of electrons in heterolytic bond cleavage. In this usage, a leaving group is a less formal but more commonly used synonym of the term nucleofuge. In this context, leaving groups are generally anions or neutral species, departing from neutral or cationic substrates, respectively, though in rare cases, cations leaving from a dicationic substrate are also known.
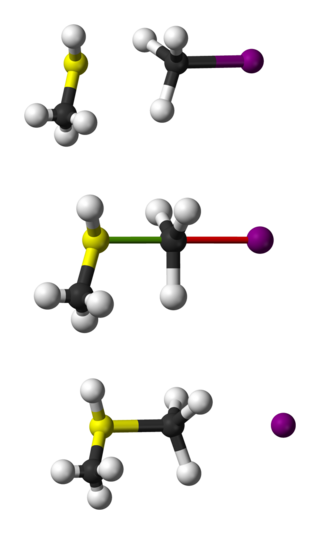
Bimolecular nucleophilic substitution (SN2) is a type of reaction mechanism that is common in organic chemistry. In the SN2 reaction, a strong nucleophile forms a new bond to an sp3-hybridised carbon atom via a backside attack, all while the leaving group detaches from the reaction center in a concerted fashion.

An aldol condensation is a condensation reaction in organic chemistry in which two carbonyl moieties react to form a β-hydroxyaldehyde or β-hydroxyketone, and this is then followed by dehydration to give a conjugated enone.
In organic chemistry, a carbanion is an anion in which carbon is negatively charged.
In physical organic chemistry, a kinetic isotope effect (KIE) is the change in the reaction rate of a chemical reaction when one of the atoms in the reactants is replaced by one of its isotopes. Formally, it is the ratio of rate constants for the reactions involving the light (kL) and the heavy (kH) isotopically substituted reactants (isotopologues):
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912.

Hammond's postulate, is a hypothesis in physical organic chemistry which describes the geometric structure of the transition state in an organic chemical reaction. First proposed by George Hammond in 1955, the postulate states that:
If two states, as, for example, a transition state and an unstable intermediate, occur consecutively during a reaction process and have nearly the same energy content, their interconversion will involve only a small reorganization of the molecular structures.
The benzilic acid rearrangement is formally the 1,2-rearrangement of 1,2-diketones to form α-hydroxy–carboxylic acids using a base. This reaction receives its name from the reaction of benzil with potassium hydroxide to form benzilic acid. First performed by Justus von Liebig in 1838, it is the first reported example of a rearrangement reaction. It has become a classic reaction in organic synthesis and has been reviewed many times before. It can be viewed as an intramolecular redox reaction, as one carbon center is oxidized while the other is reduced.

The Darzens reaction is the chemical reaction of a ketone or aldehyde with an α-haloester in the presence of a base to form an α,β-epoxy ester, also called a "glycidic ester". This reaction was discovered by the organic chemist Auguste Georges Darzens in 1904.
In chemistry, a reaction intermediate, or intermediate, is a molecular entity arising within the sequence of a stepwise chemical reaction. It is formed as the reaction product of an elementary step, from the reactants and/or preceding intermediates, but is consumed in a later step. It does not appear in the chemical equation for the overall reaction.
The Kornblum–DeLaMare rearrangement is a rearrangement reaction in organic chemistry in which a primary or secondary organic peroxide is converted to the corresponding ketone and alcohol under acid or base catalysis. The reaction is relevant as a tool in organic synthesis and is a key step in the biosynthesis of prostaglandins.
Oxidative decarboxylation is a decarboxylation reaction caused by oxidation. Most are accompanied by α- Ketoglutarate α- Decarboxylation caused by dehydrogenation of hydroxyl carboxylic acids such as carbonyl carboxylic acid, malic acid, isocitric acid, etc.
The Wallach rearrangement, also named Wallach transformation, is a name reaction in the organic chemistry. It is named after Otto Wallach, who discovered this reaction in 1880. In general it is a strong acid-promoted conversion of azoxybenzenes into hydroxyazobenzenes.
In organic chemistry, the Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, is a special type of elimination reaction in which two vicinal (adjacent) substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents, unlike regular eliminations, which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
More O’Ferrall–Jencks plots are two-dimensional representations of multiple reaction coordinate potential energy surfaces for chemical reactions that involve simultaneous changes in two bonds. As such, they are a useful tool to explain or predict how changes in the reactants or reaction conditions can affect the position and geometry of the transition state of a reaction for which there are possible competing pathways.
The Evelyn effect is defined as the phenomena in which the product ratios in a chemical reaction change as the reaction proceeds. This phenomenon contradicts the fundamental principle in organic chemistry by reactions always go by the lowest energy pathway. The favored product should remain so throughout a reaction run at constant conditions. However, the ratio of alkenes before the synthesis is complete shows that the favored product to is not the favored product. The basic idea here is that the proportions of the various alkene products changes as a function of time with a change in mechanism.
Ether cleavage refers to chemical substitution reactions that lead to the cleavage of ethers. Due to the high chemical stability of ethers, the cleavage of the C-O bond is uncommon in the absence of specialized reagents or under extreme conditions.