Epilepsy is a group of non-communicable neurological disorders characterized by recurrent epileptic seizures. An epileptic seizure is the clinical manifestation of an abnormal, excessive, and synchronized electrical discharge in the brain cells called neurons. The occurrence of two or more unprovoked seizures defines epilepsy. The occurrence of just one seizure may warrant the definition in a more clinical usage where recurrence may be able to be prejudged. Epileptic seizures can vary from brief and nearly undetectable periods to long periods of vigorous shaking due to abnormal electrical activity in the brain. These episodes can result in physical injuries, either directly such as broken bones or through causing accidents. In epilepsy, seizures tend to recur and may have no immediate underlying cause. Isolated seizures that are provoked by a specific cause such as poisoning are not deemed to represent epilepsy. People with epilepsy may be treated differently in various areas of the world and experience varying degrees of social stigma due to the alarming nature of their symptoms.
Anticonvulsants are a diverse group of pharmacological agents used in the treatment of epileptic seizures. Anticonvulsants are also increasingly being used in the treatment of bipolar disorder and borderline personality disorder, since many seem to act as mood stabilizers, and for the treatment of neuropathic pain. Anticonvulsants suppress the excessive rapid firing of neurons during seizures. Anticonvulsants also prevent the spread of the seizure within the brain.

Oxcarbazepine, sold under the brand name Trileptal among others, is a medication used to treat epilepsy. For epilepsy it is used for both focal seizures and generalized seizures. It has been used both alone and as add-on therapy in people with bipolar disorder who have had no success with other treatments. It is taken by mouth.
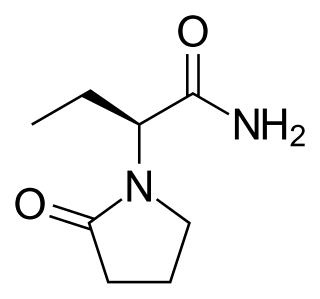
Levetiracetam, sold under the brand name Keppra among others, is a medication used to treat epilepsy. It is used for partial-onset, myoclonic, or tonic–clonic seizures and is taken either by mouth as an immediate or extended release formulation or by injection into a vein.
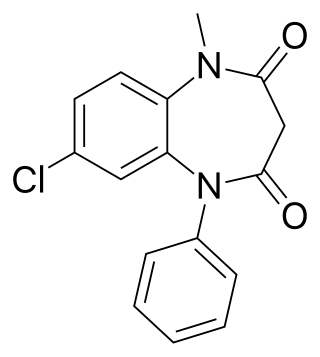
Clobazam, sold under the brand names Frisium, Onfi and others, is a benzodiazepine class medication that was patented in 1968. Clobazam was first synthesized in 1966 and first published in 1969. Clobazam was originally marketed as an anxioselective anxiolytic since 1970, and an anticonvulsant since 1984. The primary drug-development goal was to provide greater anxiolytic, anti-obsessive efficacy with fewer benzodiazepine-related side effects.

Ethosuximide, sold under the brand name Zarontin among others, is a medication used to treat absence seizures. It may be used by itself or with other antiseizure medications such as valproic acid. Ethosuximide is taken by mouth.
T-type calcium channels are low voltage activated calcium channels that become inactivated during cell membrane hyperpolarization but then open to depolarization. The entry of calcium into various cells has many different physiological responses associated with it. Within cardiac muscle cell and smooth muscle cells voltage-gated calcium channel activation initiates contraction directly by allowing the cytosolic concentration to increase. Not only are T-type calcium channels known to be present within cardiac and smooth muscle, but they also are present in many neuronal cells within the central nervous system. Different experimental studies within the 1970s allowed for the distinction of T-type calcium channels from the already well-known L-type calcium channels. The new T-type channels were much different from the L-type calcium channels due to their ability to be activated by more negative membrane potentials, had small single channel conductance, and also were unresponsive to calcium antagonist drugs that were present. These distinct calcium channels are generally located within the brain, peripheral nervous system, heart, smooth muscle, bone, and endocrine system.

Saclofen is a competitive antagonist for the GABAB receptor. This drug is an analogue of the GABAB agonist baclofen. The GABAB receptor is heptahelical receptor, expressed as an obligate heterodimer, which couples to the Gi/o class of heterotrimeric G-proteins. The action of saclofen on the central nervous system is understandably modest, because G-proteins rely on an enzyme cascade to alter cell behavior while ionotropic receptors immediately change the ionic permeability of the neuronal plasma membrane, thus changing its firing patterns. These particular receptors, presynaptically inhibit N- and P/Q- voltage-gated calcium channels (VGCCs) via a direct interaction of the dissociated beta gamma subunit of the g-protein with the intracellular loop between the 1st and 2nd domain of the VGCC's alpha-subunit; postsynaptically, these potentiate Kir currents. Both result in inhibitory effects.

Seletracetam is a pyrrolidone-derived drug of the racetam family that is structurally related to levetiracetam. It was under development by UCB Pharmaceuticals as a more potent and effective anticonvulsant drug to replace levetiracetam but its development has been halted.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).

Loreclezole is a sedative and an anticonvulsant which acts as a GABAA receptor positive allosteric modulator. The binding site of loreclezole has been shown experimentally to be shared by valerenic acid, an extract of the root of the valerian plant. Structurally, loreclezole is a triazole derivative. In animal seizure models, loreclezole is protective against pentylenetetrazol seizures but is less active in the maximal electroshock test. In addition, at low, nontoxic doses, the drug has anti-absence activity in a genetic model of generalized absence epilepsy. Consequently, loreclezole has a profile of activity similar to that of benzodiazepines. A potential benzodiazepine-like interaction with GABA receptors is suggested by the observation that the anticonvulsant effects of loreclezole can be reversed by benzodiazepine receptor inverse agonists. The benzodiazepine antagonist flumazenil, however, fails to alter the anticonvulsant activity of loreclezole, indicating that loreclezole is not a benzodiazepine receptor agonist. Using native rat and cloned human GABA-A receptors, loreclezole strongly potentiated GABA-activated chloride current. However, activity of the drug did not require the presence of the γ-subunit and was not blocked by flumazenil, confirming that loreclezole does not interact with the benzodiazepine recognition site.

Lacosamide, sold under the brand name Vimpat among others, is a medication used for the treatment of partial-onset seizures and primary generalized tonic-clonic seizures. It is used by mouth or intravenously.
Post-traumatic epilepsy (PTE) is a form of acquired epilepsy that results from brain damage caused by physical trauma to the brain. A person with PTE experiences repeated post-traumatic seizures more than a week after the initial injury. PTE is estimated to constitute 5% of all cases of epilepsy and over 20% of cases of acquired epilepsy.
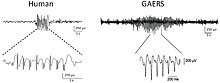
Spike-and-wave is a pattern of the electroencephalogram (EEG) typically observed during epileptic seizures. A spike-and-wave discharge is a regular, symmetrical, generalized EEG pattern seen particularly during absence epilepsy, also known as ‘petit mal’ epilepsy. The basic mechanisms underlying these patterns are complex and involve part of the cerebral cortex, the thalamocortical network, and intrinsic neuronal mechanisms.
Epileptogenesis is the gradual process by which a typical brain develops epilepsy. Epilepsy is a chronic condition in which seizures occur. These changes to the brain occasionally cause neurons to fire in an abnormal, hypersynchronous manner, known as a seizure.

Metapramine is a tricyclic antidepressant (TCA) developed by Rhone Poulenc that was introduced for the treatment of depression in France in 1984. In addition to its efficacy against affective disorders, it also has analgesic properties, and may be useful in the treatment of pain.
Jeavons syndrome is a type of epilepsy. It is one of the most distinctive reflex syndromes of idiopathic generalized epilepsy characterized by the triad of eyelid myoclonia with and without absences, eye-closure-induced seizures, EEG paroxysms, or both, and photosensitivity. Eyelid myoclonia with or without absences is a form of epileptic seizure manifesting with myoclonic jerks of the eyelids with or without a brief absence. These are mainly precipitated by closing of the eyes and lights. Eyelid myoclonia is the defining seizure type of Jeavons syndrome.

Methylazoxymethanol acetate, MAM, is a neurotoxin which reduces DNA synthesis used in making animal models of neurological diseases including schizophrenia and epilepsy. MAM is found in cycad seeds, and causes zamia staggers. It selectively targets neuroblasts in the central nervous system. In rats, administration of MAM affects structures in the brain which are developing most quickly. It is an acetate of methylazoxymethanol.
Animal models of epilepsy have helped to advance the understanding of how normal brains develop epilepsy, and have been used in pre-clinical trials of antiepileptic drugs. Epilepsy is a set of syndromes which have in common a predisposition to recurrent epileptic seizures. Animal models of epilepsy and seizures can be divided into three basic categories: genetic animal models, chemically induced models, and electrically induced models. New models are using light-gated ion channels to turn on cell firing and these are part of optogenetic induction models of epilepsy.
Gene therapy is being studied for some forms of epilepsy. It relies on viral or non-viral vectors to deliver DNA or RNA to target brain areas where seizures arise, in order to prevent the development of epilepsy or to reduce the frequency and/or severity of seizures. Gene therapy has delivered promising results in early stage clinical trials for other neurological disorders such as Parkinson's disease, raising the hope that it will become a treatment for intractable epilepsy.