Related Research Articles
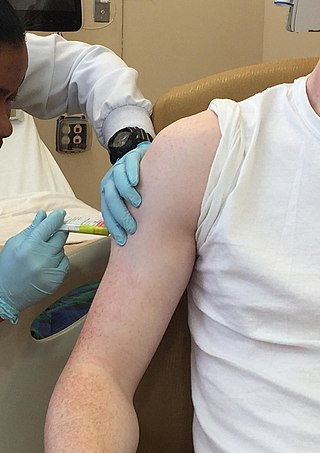
Clinical trials are prospective biomedical or behavioral research studies on human participants designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.

Current good manufacturing practices (cGMP) are those conforming to the guidelines recommended by relevant agencies. Those agencies control the authorization and licensing of the manufacture and sale of food and beverages, cosmetics, pharmaceutical products, dietary supplements, and medical devices. These guidelines provide minimum requirements that a manufacturer must meet to assure that their products are consistently high in quality, from batch to batch, for their intended use. The rules that govern each industry may differ significantly; however, the main purpose of GMP is always to prevent harm from occurring to the end user. Additional tenets include ensuring the end product is free from contamination, that it is consistent in its manufacture, that its manufacture has been well documented, that personnel are well trained, and that the product has been checked for quality more than just at the end phase. GMP is typically ensured through the effective use of a quality management system (QMS).

The United States Food and Drug Administration's Investigational New Drug (IND) program is the means by which a pharmaceutical company obtains permission to start human clinical trials and to ship an experimental drug across state lines before a marketing application for the drug has been approved. Regulations are primarily at 21 CFR 312. Similar procedures are followed in the European Union, Japan, and Canada.
Clinical monitoring is the oversight and administrative efforts that monitor a participant's health and efficacy of the treatment during a clinical trial. Both independent and government-run grant-funding agencies, such as the National Institutes of Health (NIH) and the World Health Organization (WHO), require data and safety monitoring protocols for Phase I and II clinical trials conforming to their standards.
An adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product.
A serious adverse event (SAE) in human drug trials is defined as any untoward medical occurrence that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage

Medical research, also known as experimental medicine, encompasses a wide array of research, extending from "basic research", – involving fundamental scientific principles that may apply to a preclinical understanding – to clinical research, which involves studies of people who may be subjects in clinical trials. Within this spectrum is applied research, or translational research, conducted to expand knowledge in the field of medicine.
Acquisition or collection of clinical trial data can be achieved through various methods that may include, but are not limited to, any of the following: paper or electronic medical records, paper forms completed at a site, interactive voice response systems, local electronic data capture systems, or central web based systems.
The Declaration of Helsinki is a set of ethical principles regarding human experimentation developed originally in 1964 for the medical community by the World Medical Association (WMA). It is widely regarded as the cornerstone document on human research ethics.
In the experimental (non-clinical) research arena, good laboratory practice or GLP is a quality system of management controls for research laboratories and organizations to ensure the uniformity, consistency, reliability, reproducibility, quality, and integrity of products in development for human or animal health through non-clinical safety tests; from physio-chemical properties through acute to chronic toxicity tests.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.

EudraLex is the collection of rules and regulations governing medicinal products in the European Union.
The Good Clinical Practice Directive lays down principles and detailed guidelines for good clinical practice as regards conducting clinical trials of medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products.
In natural and social science research, a protocol is most commonly a predefined procedural method in the design and implementation of an experiment. Protocols are written whenever it is desirable to standardize a laboratory method to ensure successful replication of results by others in the same laboratory or by other laboratories. Additionally, and by extension, protocols have the advantage of facilitating the assessment of experimental results through peer review. In addition to detailed procedures, equipment, and instruments, protocols will also contain study objectives, reasoning for experimental design, reasoning for chosen sample sizes, safety precautions, and how results were calculated and reported, including statistical analysis and any rules for predefining and documenting excluded data to avoid bias.
A Clinical Research Coordinator (CRC) is a person responsible for conducting clinical trials using good clinical practice (GCP) under the auspices of a Principal Investigator (PI).
In health care, a clinical trial is a comparison test of a medication or other medical treatment, versus a placebo, other medications or devices, or the standard medical treatment for a patient's condition.
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:
In order to comply with government regulatory requirements pertinent to clinical trials, every organization involved in clinical trials must maintain and store certain documents, images and content related to the clinical trial. Depending on the regulatory jurisdiction, this information may be stored in the trial master file or TMF, which today takes the form of an electronic trial master file (eTMF). The International Conference on Harmonization (ICH) published a consolidated guidance for industry on Good Clinical Practice in 1996 with the objective of providing a unified standard for the European Union, Japan, and the United States of America to facilitate mutual acceptance of clinical data by the regulatory authorities in those jurisdictions. This guidance document established the requirement across all ICH regions to establish trial master files containing essential documents that individually and collectively permit evaluation of the conduct of a trial and the quality of the data produced.[2] In some jurisdictions, for example the USA, there is no specific requirement for a trial master file. However, if the regulatory authority requires ICH GCP to be followed, then there is consequently a requirement to create and maintain a trial master file.[2]
Guidances for statistics in regulatory affairs refers to specific documents or guidelines that provide instructions, recommendations, and standards pertaining to the application of statistical methodologies and practices within the regulatory framework of industries such as pharmaceuticals and medical devices. These guidances serve as a reference for statisticians, researchers, and professionals involved in designing, conducting, analyzing, and reporting studies and trials in compliance with regulatory requirements. These documents embody the prevailing perspectives of regulatory agencies on specific subjects. It is worth noting that in the United States, the term "Guidances" is used, while in Europe, the term "Guidelines" is employed.
References
- ↑ "ICH Official web site : ICH". www.ich.org. Retrieved 2021-11-24.
- ↑ https://health.ec.europa.eu/medicinal-products/clinical-trials/clinical-trials-directive-200120ec_en
- ↑ Commissioner, Office of the. "Clinical Trials and Human Subject Protection". www.fda.gov. Retrieved 2018-11-01.
- ↑ "Good Clinical Practice Training | grants.nih.gov". grants.nih.gov. Retrieved 2020-04-03.
- ↑ Kimmelman, Jonathan; Weijer, Charles; Meslin, Eric M (2009). "Helsinki discords: FDA, ethics, and international drug trials". The Lancet. 373 (9657): 13–14. doi:10.1016/S0140-6736(08)61936-4. PMID 19121708.
- ↑ Ben Goldacre (2012). Bad Pharma. London: Fourth Estate. OL 25682902M.