Related Research Articles
Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.

Wilkinson's catalyst is the common name for chloridotris(triphenylphosphine)rhodium(I), a coordination complex of rhodium with the formula [RhCl(PPh3)3], where 'Ph' denotes a phenyl group). It is a red-brown colored solid that is soluble in hydrocarbon solvents such as benzene, and more so in tetrahydrofuran or chlorinated solvents such as dichloromethane. The compound is widely used as a catalyst for hydrogenation of alkenes. It is named after chemist and Nobel laureate Sir Geoffrey Wilkinson, who first popularized its use.
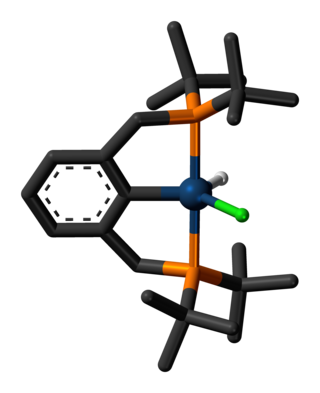
In chemistry, a transition metal pincer complex is a type of coordination complex with a pincer ligand. Pincer ligands are chelating agents that binds tightly to three adjacent coplanar sites in a meridional configuration. The inflexibility of the pincer-metal interaction confers high thermal stability to the resulting complexes. This stability is in part ascribed to the constrained geometry of the pincer, which inhibits cyclometallation of the organic substituents on the donor sites at each end. In the absence of this effect, cyclometallation is often a significant deactivation process for complexes, in particular limiting their ability to effect C-H bond activation. The organic substituents also define a hydrophobic pocket around the reactive coordination site. Stoichiometric and catalytic applications of pincer complexes have been studied at an accelerating pace since the mid-1970s. Most pincer ligands contain phosphines. Reactions of metal-pincer complexes are localized at three sites perpendicular to the plane of the pincer ligand, although in some cases one arm is hemi-labile and an additional coordination site is generated transiently. Early examples of pincer ligands were anionic with a carbanion as the central donor site and flanking phosphine donors; these compounds are referred to as PCP pincers.

The Pauson–Khand (PK) reaction is a chemical reaction, described as a [2+2+1] cycloaddition. In it, an alkyne, an alkene and carbon monoxide combine into a α,β-cyclopentenone in the presence of a metal-carbonyl catalyst.

Dicobalt octacarbonyl is an organocobalt compound with composition Co2(CO)8. This metal carbonyl is used as a reagent and catalyst in organometallic chemistry and organic synthesis, and is central to much known organocobalt chemistry. It is the parent member of a family of hydroformylation catalysts. Each molecule consists of two cobalt atoms bound to eight carbon monoxide ligands, although multiple structural isomers are known. Some of the carbonyl ligands are labile.
In chemistry, the Noyori asymmetric hydrogenation refers to methodology for enantioselective reduction of ketones and related functional groups. This methodology was introduced by Ryoji Noyori, who shared the Nobel Prize in Chemistry in 2001 for contributions to asymmetric hydrogenation. These hydrogenations are used in the production of several drugs, such as the antibacterial levofloxin, the antibiotic carbapenem, and the antipsychotic agent BMS181100.
Bis(oxazoline) ligands (often abbreviated BOX ligands) are a class of privileged chiral ligands containing two oxazoline rings. They are typically C2‑symmetric and exist in a wide variety of forms; with structures based around CH2 or pyridine linkers being particularly common (often generalised BOX and PyBOX respectively). The coordination complexes of bis(oxazoline) ligands are used in asymmetric catalysis. These ligands are examples of C2-symmetric ligands.
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being awarded one half of the 2001 Nobel Prize in Chemistry.

Oxazoline is a five-membered heterocyclic organic compound with the formula C3H5NO. It is the parent of a family of compounds called oxazolines, which contain non-hydrogenic substituents on carbon and/or nitrogen. Oxazolines are the unsaturated analogues of oxazolidines, and they are isomeric with isoxazolines, where the N and O are directly bonded. Two isomers of oxazoline are known, depending on the location of the double bond.
Organogold chemistry is the study of compounds containing gold–carbon bonds. They are studied in academic research, but have not received widespread use otherwise. The dominant oxidation states for organogold compounds are I with coordination number 2 and a linear molecular geometry and III with CN = 4 and a square planar molecular geometry.

Organorhodium chemistry is the chemistry of organometallic compounds containing a rhodium-carbon chemical bond, and the study of rhodium and rhodium compounds as catalysts in organic reactions.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
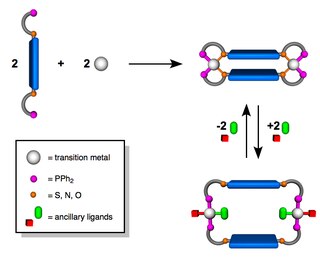
The Weak-Link Approach (WLA) is a supramolecular coordination-based assembly methodology, first introduced in 1998 by the Mirkin Group at Northwestern University. This method takes advantage of hemilabile ligands -ligands that contain both strong and weak binding moieties- that can coordinate to metal centers and quantitatively assemble into a single condensed ‘closed’ structure. Unlike other supramolecular assembly methods, the WLA allows for the synthesis of supramolecular complexes that can be modulated from rigid ‘closed’ structures to flexible ‘open’ structures through reversible binding of allosteric effectors at the structural metal centers. The approach is general and has been applied to a variety of metal centers and ligand designs including those with utility in catalysis and allosteric regulation.
In Lewis acid catalysis of organic reactions, a metal-based Lewis acid acts as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.

Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Their complexes are particularly effective at generating single enatiomers in reactions involving highly symmetric transition states, such as allylic substitutions, which are typically difficult to perform stereoselectively. The ligands are bidentate and have been shown to be hemilabile with the softer P‑donor being more firmly bound than the harder N‑donor.

Trisoxazolines are a class of tridentate, chiral ligands composed of three oxazoline rings. Despite being neutral they are able to form stable complexes with high oxidation state metals, such as rare earths, due to the chelate effect. The ligands have been investigated for molecular recognition and their complexes are used in asymmetric catalysts and polymerisation.
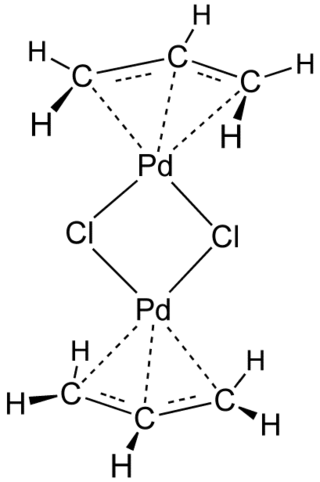
Transition-metal allyl complexes are coordination complexes with allyl and its derivatives as ligands. Allyl is the radical with the connectivity CH2CHCH2, although as a ligand it is usually viewed as an allyl anion CH2=CH−CH2−, which is usually described as two equivalent resonance structures.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.
In homogeneous catalysis, C2-symmetric ligands refer to ligands that lack mirror symmetry but have C2 symmetry. Such ligands are usually bidentate and are valuable in catalysis. The C2 symmetry of ligands limits the number of possible reaction pathways and thereby increases enantioselectivity, relative to asymmetrical analogues. C2-symmetric ligands are a subset of chiral ligands. Chiral ligands, including C2-symmetric ligands, combine with metals or other groups to form chiral catalysts. These catalysts engage in enantioselective chemical synthesis, in which chirality in the catalyst yields chirality in the reaction product.

Water oxidation catalysis (WOC) is the acceleration (catalysis) of the conversion of water into oxygen and protons:
References
- ↑ Bader, Armin; Lindner, Ekkehard (April 1991). "Coordination chemistry and catalysis with hemilabile oxygen-phosphorus ligands". Coordination Chemistry Reviews. 108 (1): 27–110. doi:10.1016/0010-8545(91)80013-4.
- ↑ Braunstein, Pierre; Naud, Frédéric (16 February 2001). "Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems". Angewandte Chemie International Edition. 40 (4): 680–699. doi: 10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0 . PMID 11241595.
- ↑ Slone, Caroline S.; Weinberger, Dana A.; Mirkin, Chad A. (1999), "The Transition Metal Coordination Chemistry of Hemilabile Ligands", Progress in Inorganic Chemistry, John Wiley & Sons, Ltd, pp. 233–350, doi:10.1002/9780470166499.ch3, ISBN 978-0-470-16649-9 , retrieved 2021-04-02
- ↑ Braunstein, Pierre; Naud, Frédéric (2001). "Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems". Angewandte Chemie International Edition. 40 (4): 680–699. doi: 10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0 . ISSN 1521-3773. PMID 11241595.
- ↑ Miller, Eileen M.; Shaw, Bernard L. (1 January 1974). "Kinetic and other studies on oxidative addition reactions of iridium phosphine complexes of the type trans-[IrCl(CO)(PMe2R)2](R = Ph, o-MeO·C6H4, or p-MeO·C6H4)". Journal of the Chemical Society, Dalton Transactions (5): 480–485. doi:10.1039/DT9740000480.
- ↑ Nomura, Nobuyoshi; Jin, Jian; Park, Haengsoon; RajanBabu, T. V. (1 January 1998). "The Hydrovinylation Reaction: A New Highly Selective Protocol Amenable to Asymmetric Catalysis". Journal of the American Chemical Society. 120 (2): 459–460. doi:10.1021/ja973548n.
- ↑ RajanBabu, T. V. (1 August 2003). "Asymmetric Hydrovinylation Reaction". Chemical Reviews. 103 (8): 2845–2860. doi:10.1021/cr020040g. PMID 12914483.
- ↑ Verdaguer, Xavier; Moyano, Albert; Pericàs, Miquel A.; Riera, Antoni; Maestro, Miguel Angel; Mahía, José (1 October 2000). "A New Chiral Bidentate (P,S) Ligand for the Asymmetric Intermolecular Pauson−Khand Reaction". Journal of the American Chemical Society. 122 (41): 10242–10243. doi:10.1021/ja001839h.
- ↑ Jiménez, M. Victoria; Fernández-Tornos, Javier; Pérez-Torrente, Jesús J.; Modrego, Francisco J.; Winterle, Sonja; Cunchillos, Carmen; Lahoz, Fernando J.; Oro, Luis A. (24 October 2011). "Iridium(I) Complexes with Hemilabile N-Heterocyclic Carbenes: Efficient and Versatile Transfer Hydrogenation Catalysts" (PDF). Organometallics. 30 (20): 5493–5508. doi:10.1021/om200747k. hdl:10261/57986.