
Zellweger syndrome is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes in the cells of an individual. It is one of a family of disorders called Zellweger spectrum disorders which are leukodystrophies. Zellweger syndrome is named after Hans Zellweger (1909–1990), a Swiss-American pediatrician, a professor of pediatrics and genetics at the University of Iowa who researched this disorder.
Phytol is an acyclic hydrogenated diterpene alcohol that is used as a precursor for the manufacture of synthetic forms of vitamin E and vitamin K1, as well as in the fragrance industry. Its other commercial uses include cosmetics, shampoos, toilet soaps, and detergents, as well as in some cannabis distillates as a diluent or for flavoring. Its worldwide use has been estimated to be approximately 0.1–1.0 metric tons per year.
Refsum disease is an autosomal recessive neurological disease that results in the over-accumulation of phytanic acid in cells and tissues. It is one of several disorders named after Norwegian neurologist Sigvald Bernhard Refsum (1907–1991). Refsum disease typically is adolescent onset and is diagnosed by above average levels of phytanic acid. Humans obtain the necessary phytanic acid primarily through diet. It is still unclear what function phytanic acid plays physiologically in humans, but has been found to regulate fatty acid metabolism in the liver of mice.
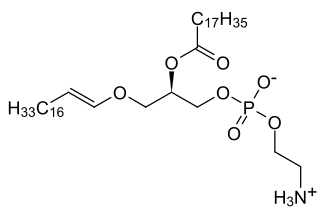
Phytanic acid is a branched chain fatty acid that humans can obtain through the consumption of dairy products, ruminant animal fats, and certain fish. Western diets are estimated to provide 50–100 mg of phytanic acid per day. In a study conducted in Oxford, individuals who consumed meat had, on average, a 6.7-fold higher geometric mean plasma phytanic acid concentration than did vegans.
Peroxisomal disorders represent a class of medical conditions caused by defects in peroxisome functions. This may be due to defects in single enzymes important for peroxisome function or in peroxins, proteins encoded by PEX genes that are critical for normal peroxisome assembly and biogenesis.
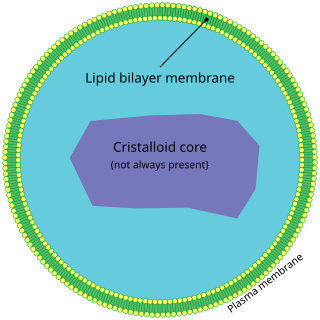
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by abnormally short arms and legs (rhizomelia), seizures, recurrent respiratory tract infections and congenital cataracts.

Pipecolic acidemia is a very rare autosomal recessive metabolic disorder that is caused by a peroxisomal defect.
D-Bifunctional protein deficiency is an autosomal recessive peroxisomal fatty acid oxidation disorder. Peroxisomal disorders are usually caused by a combination of peroxisomal assembly defects or by deficiencies of specific peroxisomal enzymes. The peroxisome is an organelle in the cell similar to the lysosome that functions to detoxify the cell. Peroxisomes contain many different enzymes, such as catalase, and their main function is to neutralize free radicals and detoxify drugs. For this reason peroxisomes are ubiquitous in the liver and kidney. D-BP deficiency is the most severe peroxisomal disorder, often resembling Zellweger syndrome.
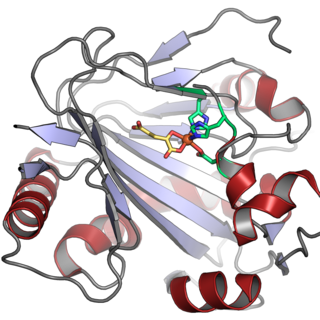
In enzymology, a phytanoyl-CoA dioxygenase (EC 1.14.11.18) is an enzyme that catalyzes the chemical reaction

Peroxisomal targeting signal 1 receptor (PTS1R) is a protein that in humans is encoded by the PEX5 gene.

Peroxisome biogenesis factor 1, also known as PEX1, is a protein which in humans is encoded by the PEX1 gene.
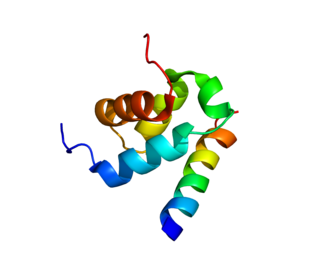
Peroxisomal membrane protein PEX14 is a protein that in humans is encoded by the PEX14 gene.

ATP-binding cassette sub-family D member 3 is a protein that in humans is encoded by the ABCD3 gene.

Peroxisomal biogenesis factor 2 is a protein that in humans is encoded by the PEX2 gene.

Peroxisome assembly protein 12 is a protein that in humans is encoded by the PEX12 gene.

Peroxisome assembly factor 2 is a protein that in humans is encoded by the PEX6 gene. PEX6 is an AAA ATPase that localizes to the peroxisome. PEX6 forms a hexamer with PEX1 and is recruited to the membrane by PEX26.
Peroxisome biogenesis factor 10 is a protein that in humans is encoded by the PEX10 gene. Alternative splicing results in two transcript variants encoding different isoforms.

Peroxisomal membrane protein PEX16 is a protein that in humans is encoded by the PEX16 gene.

Peroxisome assembly protein 26 is a protein that in humans is encoded by the PEX26 gene.
Zellweger spectrum disorders are a group of rare disorders that create the same disease process. The subdivisions of this spectrum are hyperpipecolic acidemia, infantile Refsum disease, neonatal adrenoleukodystrophy, and Zellweger syndrome. It can also be referred to as peroxisomal biogenesis disorders, Zellweger syndrome spectrum, NALD, cerebrohepatorenal syndrome, and ZSS. It can affect many body organs, including the kidneys, eyes, and hearing. It is named after Hans Zellweger.