
Fick's laws of diffusion describe diffusion and were first posited by Adolf Fick in 1855 on the basis of largely experimental results. They can be used to solve for the diffusion coefficient, D. Fick's first law can be used to derive his second law which in turn is identical to the diffusion equation.
The kinetic theory of gases is a simple classical model of the thermodynamic behavior of gases. It treats a gas as composed of numerous particles, too small to see with a microscope, which are constantly in random motion. Their collisions with each other and with the walls of their container are used to explain physical properties of the gas—for example the relationship between its temperature, pressure and volume. The particles are now known to be the atoms or molecules of the gas.
Physisorption, also called physical adsorption, is a process in which the electronic structure of the atom or molecule is barely perturbed upon adsorption.

In probability theory and statistics, the gamma distribution is a versatile two-parameter family of continuous probability distributions. The exponential distribution, Erlang distribution, and chi-squared distribution are special cases of the gamma distribution. There are two equivalent parameterizations in common use:
- With a shape parameter k and a scale parameter θ
- With a shape parameter and an inverse scale parameter , called a rate parameter.
Adsorption is the adhesion of atoms, ions or molecules from a gas, liquid or dissolved solid to a surface. This process creates a film of the adsorbate on the surface of the adsorbent. This process differs from absorption, in which a fluid is dissolved by or permeates a liquid or solid. While adsorption does often precede absorption, which involves the transfer of the absorbate into the volume of the absorbent material, alternatively, adsorption is distinctly a surface phenomenon, wherein the adsorbate does not penetrate through the material surface and into the bulk of the adsorbent. The term sorption encompasses both adsorption and absorption, and desorption is the reverse of sorption.
Temperature programmed desorption (TPD) is the method of observing desorbed molecules from a surface when the surface temperature is increased. When experiments are performed using well-defined surfaces of single-crystalline samples in a continuously pumped ultra-high vacuum (UHV) chamber, then this experimental technique is often also referred to as thermal desorption spectroscopy or thermal desorption spectrometry (TDS).
Desorption is the physical process where adsorbed atoms or molecules are released from a surface into the surrounding vacuum or fluid. This occurs when a molecule gains enough energy to overcome the activation barrier and the binding energy that keep it attached to the surface.

Wetting is the ability of a liquid to maintain contact with a solid surface, resulting from intermolecular interactions when the two are brought together. This happens in presence of a gaseous phase or another liquid phase not miscible with the first one. The degree of wetting (wettability) is determined by a force balance between adhesive and cohesive forces. There are two types of wetting: non-reactive wetting and reactive wetting.
Brunauer–Emmett–Teller (BET) theory aims to explain the physical adsorption of gas molecules on a solid surface and serves as the basis for an important analysis technique for the measurement of the specific surface area of materials. The observations are very often referred to as physical adsorption or physisorption. In 1938, Stephen Brunauer, Paul Hugh Emmett, and Edward Teller presented their theory in the Journal of the American Chemical Society. BET theory applies to systems of multilayer adsorption that usually utilizes a probing gas (called the adsorbate) that does not react chemically with the adsorptive (the material upon which the gas attaches to) to quantify specific surface area. Nitrogen is the most commonly employed gaseous adsorbate for probing surface(s). For this reason, standard BET analysis is most often conducted at the boiling temperature of N2 (77 K). Other probing adsorbates are also utilized, albeit less often, allowing the measurement of surface area at different temperatures and measurement scales. These include argon, carbon dioxide, and water. Specific surface area is a scale-dependent property, with no single true value of specific surface area definable, and thus quantities of specific surface area determined through BET theory may depend on the adsorbate molecule utilized and its adsorption cross section.
The sticking probability is the probability that molecules are trapped on surfaces and adsorb chemically. From Langmuir's adsorption isotherm, molecules cannot adsorb on surfaces when the adsorption sites are already occupied by other molecules, so the sticking probability can be expressed as follows:
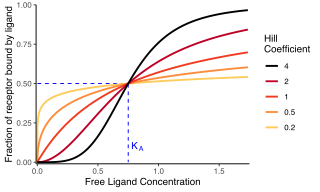
In biochemistry and pharmacology, the Hill equation refers to two closely related equations that reflect the binding of ligands to macromolecules, as a function of the ligand concentration. A ligand is "a substance that forms a complex with a biomolecule to serve a biological purpose", and a macromolecule is a very large molecule, such as a protein, with a complex structure of components. Protein-ligand binding typically changes the structure of the target protein, thereby changing its function in a cell.
The principle of detailed balance can be used in kinetic systems which are decomposed into elementary processes. It states that at equilibrium, each elementary process is in equilibrium with its reverse process.
Reactions on surfaces are reactions in which at least one of the steps of the reaction mechanism is the adsorption of one or more reactants. The mechanisms for these reactions, and the rate equations are of extreme importance for heterogeneous catalysis. Via scanning tunneling microscopy, it is possible to observe reactions at the solid gas interface in real space, if the time scale of the reaction is in the correct range. Reactions at the solid–gas interface are in some cases related to catalysis.

A pendulum is a body suspended from a fixed support so that it swings freely back and forth under the influence of gravity. When a pendulum is displaced sideways from its resting, equilibrium position, it is subject to a restoring force due to gravity that will accelerate it back towards the equilibrium position. When released, the restoring force acting on the pendulum's mass causes it to oscillate about the equilibrium position, swinging it back and forth. The mathematics of pendulums are in general quite complicated. Simplifying assumptions can be made, which in the case of a simple pendulum allow the equations of motion to be solved analytically for small-angle oscillations.
The Freundlich equation or Freundlich adsorption isotherm, an adsorption isotherm, is an empirical relationship between the quantity of a gas adsorbed into a solid surface and the gas pressure. The same relationship is also applicable for the concentration of a solute adsorbed onto the surface of a solid and the concentration of the solute in the liquid phase. In 1909, Herbert Freundlich gave an expression representing the isothermal variation of adsorption of a quantity of gas adsorbed by unit mass of solid adsorbent with gas pressure. This equation is known as Freundlich adsorption isotherm or Freundlich adsorption equation. As this relationship is entirely empirical, in the case where adsorption behavior can be properly fit by isotherms with a theoretical basis, it is usually appropriate to use such isotherms instead. The Freundlich equation is also derived (non-empirically) by attributing the change in the equilibrium constant of the binding process to the heterogeneity of the surface and the variation in the heat of adsorption.
Sticking coefficient is the term used in surface physics to describe the ratio of the number of adsorbate atoms that adsorb, or "stick", to a surface to the total number of atoms that impinge upon that surface during the same period of time. Sometimes the symbol Sc is used to denote this coefficient, and its value is between 1 and 0. The coefficient is a function of surface temperature, surface coverage (θ) and structural details as well as the kinetic energy of the impinging particles. The original formulation was for molecules adsorbing from the gas phase and the equation was later extended to adsorption from the liquid phase by comparison with molecular dynamics simulations. For use in adsorption from liquids the equation is expressed based on solute density rather than the pressure.
In materials science, segregation is the enrichment of atoms, ions, or molecules at a microscopic region in a materials system. While the terms segregation and adsorption are essentially synonymous, in practice, segregation is often used to describe the partitioning of molecular constituents to defects from solid solutions, whereas adsorption is generally used to describe such partitioning from liquids and gases to surfaces. The molecular-level segregation discussed in this article is distinct from other types of materials phenomena that are often called segregation, such as particle segregation in granular materials, and phase separation or precipitation, wherein molecules are segregated in to macroscopic regions of different compositions. Segregation has many practical consequences, ranging from the formation of soap bubbles, to microstructural engineering in materials science, to the stabilization of colloidal suspensions.
Supercritical adsorption also referred to as the adsorption of supercritical fluids, is the adsorption at above-critical temperatures. There are different tacit understandings of supercritical fluids. For example, “a fluid is considered to be ‘supercritical’ when its temperature and pressure exceed the temperature and pressure at the critical point”. In the studies of supercritical extraction, however, “supercritical fluid” is applied for a narrow temperature region of 1-1.2 or to +10 K, which is called the supercritical region.
Adsorption is the adhesion of ions or molecules onto the surface of another phase. Adsorption may occur via physisorption and chemisorption. Ions and molecules can adsorb to many types of surfaces including polymer surfaces. A polymer is a large molecule composed of repeating subunits bound together by covalent bonds. In dilute solution, polymers form globule structures. When a polymer adsorbs to a surface that it interacts favorably with, the globule is essentially squashed, and the polymer has a pancake structure.
The potential theory of Polanyi, also called Polanyi adsorption potential theory, is a model of adsorption proposed by Michael Polanyi where adsorption can be measured through the equilibrium between the chemical potential of a gas near the surface and the chemical potential of the gas from a large distance away. In this model, he assumed that the attraction largely due to Van Der Waals forces of the gas to the surface is determined by the position of the gas particle from the surface, and that the gas behaves as an ideal gas until condensation where the gas exceeds its equilibrium vapor pressure. While the adsorption theory of Henry is more applicable in low pressure and BET adsorption isotherm equation is more useful at from 0.05 to 0.35 P/Po, the Polanyi potential theory has much more application at higher P/Po (~0.1–0.8).































































































