
Louis Isaac Woolf (born 1919 in London, England; died 2021 in Vancouver, Canada) was a British biochemist who played a crucial role in the early detection (via neonatal screening) and the treatment of phenylketonuria (PKU).
Louis Isaac Woolf (born 1919 in London, England; died 2021 in Vancouver, Canada) was a British biochemist who played a crucial role in the early detection (via neonatal screening) and the treatment of phenylketonuria (PKU).
Woolf was born in London, England, on 24 April 1919. [1] He was born in Hackney, London to a Romanian Jewish family. He had 2 siblings. [2] [3]
He studied chemistry at University College London (UCL) and was awarded a PhD in 1945. [1] In 1947, he was awarded an Imperial Chemical Industries (ICI) research fellowship at the Hospital for Sick Children at Great Ormond Street, London, working on tyrosine metabolism in premature babies and inherited metabolic disorders, with a focus on amino acids. [1] [2]
Woolf believed that the metabolic disorder phenylketonuria (PKU) could be treated through dietary changes, most notably a low-phenylalanine diet. At the time, the scientific consensus was that PKU was untreatable. [4] However, the idea that it could be treated through diet was proposed by some doctors since the 1930s, shortly after the condition was first described. [5] However, this was not easy as scientists struggled to reduce the levels of phenylalanine in food. [4] Woolf's idea of using activated charcoal to filter phenylalanine from casein hydrolysate laid the groundwork for future dietary interventions, [6] which he researched in the late 1940s and early 1950s. [1]
The first successful trial of this diet involved proposing a low-phenylalanine diet as a treatment for PKU. This was done in collaboration with Horst Bickel, John W. Gerrard and other scientists in 1951. This trialled despite Woolf and others facing scepticism and professional challenges, including the belief that PKU was untreatable due to its genetic nature. The result of the trial diet on a young PKU patient led to significant clinical improvement. [7]
In 1957, Woolf and colleagues recommended mass screening for PKU using a ferric chloride test on urine samples from newborn babies. They emphasised the importance of early diagnosis and treatment, proposing screening at 21 days after birth. [1]
This urine test was the basis of the first commercial PKU screening test, Phenistix, which was released the next year. Phenistix was adopted in various locations, including the United Kingdom and the United States, where screening programmes which emphasise early detection and treatment were adopted. In 1966–1967, Woolf's screening methods were adopted in Spain, with a pilot program in Granada. [1]
Woolf moved to Vancouver in 1968, where he joined the University of British Columbia and continued research on phenylalanine biochemistry and metabolic diseases. His work extended beyond PKU to include a wide range of inborn errors of metabolism. [1]
In 1979, Woolf discussed the variants of PKU, including cases with blood concentrations of phenylalanine below typical PKU levels, and the consequences of interrupting a low-phenylalanine diet in later childhood. [1]
Woolf retired in 1984, taking the title professor emeritus. [1] He died in 2021 in Vancouver, Canada, aged 101 years old. [8]

Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
Phenylalanine is an essential α-amino acid with the formula C
9H
11NO
2. It can be viewed as a benzyl group substituted for the methyl group of alanine, or a phenyl group in place of a terminal hydrogen of alanine. This essential amino acid is classified as neutral, and nonpolar because of the inert and hydrophobic nature of the benzyl side chain. The L-isomer is used to biochemically form proteins coded for by DNA. Phenylalanine is a precursor for tyrosine, the monoamine neurotransmitters dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline), and the biological pigment melanin. It is encoded by the codons UUU and UUC.

The neonatal heel prick is a blood collection procedure done on newborns. It consists of making a pinprick puncture in one heel of the newborn to collect their blood. This technique is used frequently as the main way to collect blood from neonates. Other techniques include venous or arterial needle sticks, cord blood sampling, or umbilical line collection. This technique is often utilized for the Guthrie test, where it is used to soak the blood into pre-printed collection cards known as Guthrie cards.
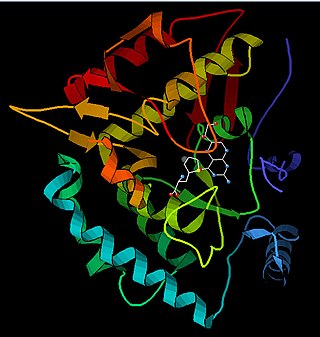
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.
Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
Ivar Asbjørn Følling was a Norwegian physician and biochemist. He first described the disease commonly known as Følling's disease or phenylketonuria (PKU).

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors, monoamine oxidase inhibitors, and tetrahydrobiopterin. Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).
Robert Guthrie, MD, Ph.D. was an American microbiologist, best known for developing the bacterial inhibition assay used to screen infants for phenylketonuria at birth, before the development of irreversible neurological damage. Guthrie also pioneered the collection of whole blood on specially designed filter paper, commonly known as "Guthrie cards" as a sample medium that could be easily collected, transported and tested. Although Guthrie is best known for developing the test for phenylketonuria, he worked tirelessly to raise awareness of the need to screen for treatable conditions and adapted his method to early screening tests for galactosemia and maple syrup urine disease.

The enzyme phenylalanine racemase is the enzyme that acts on amino acids and derivatives. It activates both the L & D stereo isomers of phenylalanine to form L-phenylalanyl adenylate and D-phenylalanyl adenylate, which are bound to the enzyme. These bound compounds are then transferred to the thiol group of the enzyme followed by conversion of its configuration, the D-isomer being the more favorable configuration of the two, with a 7 to 3 ratio between the two isomers. The racemisation reaction of phenylalanine is coupled with the highly favorable hydrolysis of adenosine triphosphate (ATP) to adenosine monophosphate (AMP) and pyrophosphate (PP), thermodynamically allowing it to proceed. This reaction is then drawn forward by further hydrolyzing PP to inorganic phosphate (Pi), via Le Chatelier's principle.
Organic acidemia is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.
Hyperphenylalaninemia is a medical condition characterized by mildly or strongly elevated concentrations of the amino acid phenylalanine in the blood. Phenylketonuria (PKU) can result in severe hyperphenylalaninemia. Phenylalanine concentrations are routinely screened in newborns by the neonatal heel prick, which takes a few drops of blood from the heel of the infant. Standard phenylalanine concentrations in unaffected persons are about 2-6mg/dl phenylalanine concentrations in those with untreated hyperphenylalaninemia can be up to 20 mg/dL. Measurable IQ deficits are often detected as phenylalanine levels approach 10 mg/dL. Phenylketonuria (PKU)-like symptoms, including more pronounced developmental defects, skin irritation, and vomiting, may appear when phenylalanine levels are near 20 mg/dL .Hyperphenylalaninemia is a recessive hereditary metabolic disorder that is caused by the body's failure to convert phenylalanine to tyrosine as a result of the entire or partial absence of the enzyme phenylalanine hydroxylase.

The history of Tay–Sachs disease started with the development and acceptance of the evolution theory of disease in the 1860s and 1870s, the possibility that science could explain and even prevent or cure illness prompted medical doctors to undertake more precise description and diagnosis of disease. Waren Tay and Bernard Sachs, two physicians of the late 19th century described the progression of the disease precisely and provided differential diagnostic criteria to distinguish it from other neurological disorders with similar symptoms.

Pegvaliase, sold under the brand name Palynziq, is a medication used for the treatment of the genetic disease phenylketonuria. It is a phenylalanine (Phe)‑metabolizing enzyme. Chemically, it is a pegylated derivative of the enzyme phenylalanine ammonia-lyase that metabolizes phenylalanine to reduce its blood levels.
The European Society for Phenylketonuria and Allied Disorders Treated as Phenylketonuria (E.S.PKU) is a Europe-based non-profit organization. It was founded in 1987 by patient-driven associations to help improve the treatment of phenylketonuria (PKU) in Europe.
Harvey Louis Levy is an American biochemical geneticist, pediatrician, physician scientist and academic. He is Senior Physician in Medicine and Genetics at Boston Children’s Hospital and Professor of Pediatrics at Harvard Medical School.

Underrepresented populations, especially black and hispanic populations with cystic fibrosis are often not successfully diagnosed. This is in part due to the minimal dissemination of existing data on patients from these underrepresented groups. While white populations do appear to experience a higher frequency of cystic fibrosis, other ethnicities are also affected and not always by the same biological mechanisms. Thus, many healthcare and treatment options are less reliable or unavailable to underrepresented populations. This issue affects the level at which public health needs are being met across the world.