Related Research Articles

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In genetics, the phenotype is the set of observable characteristics or traits of an organism. The term covers the organism's morphology, its developmental processes, its biochemical and physiological properties, its behavior, and the products of behavior. An organism's phenotype results from two basic factors: the expression of an organism's genetic code and the influence of environmental factors. Both factors may interact, further affecting the phenotype. When two or more clearly different phenotypes exist in the same population of a species, the species is called polymorphic. A well-documented example of polymorphism is Labrador Retriever coloring; while the coat color depends on many genes, it is clearly seen in the environment as yellow, black, and brown. Richard Dawkins in 1978 and then again in his 1982 book The Extended Phenotype suggested that one can regard bird nests and other built structures such as caddisfly larva cases and beaver dams as "extended phenotypes".

A DNA microarray is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles of a specific DNA sequence, known as probes. These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA sample under high-stringency conditions. Probe-target hybridization is usually detected and quantified by detection of fluorophore-, silver-, or chemiluminescence-labeled targets to determine relative abundance of nucleic acid sequences in the target. The original nucleic acid arrays were macro arrays approximately 9 cm × 12 cm and the first computerized image based analysis was published in 1981. It was invented by Patrick O. Brown. An example of its application is in SNPs arrays for polymorphisms in cardiovascular diseases, cancer, pathogens and GWAS analysis. It is also used for the identification of structural variations and the measurement of gene expression.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "candidate-gene" approach.
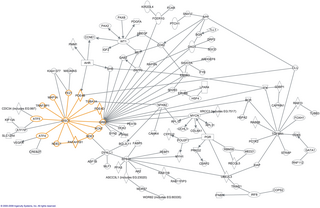
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.
Genetic architecture is the underlying genetic basis of a phenotypic trait and its variational properties. Phenotypic variation for quantitative traits is, at the most basic level, the result of the segregation of alleles at quantitative trait loci (QTL). Environmental factors and other external influences can also play a role in phenotypic variation. Genetic architecture is a broad term that can be described for any given individual based on information regarding gene and allele number, the distribution of allelic and mutational effects, and patterns of pleiotropy, dominance, and epistasis.

Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.
Biomedical text mining refers to the methods and study of how text mining may be applied to texts and literature of the biomedical domain. As a field of research, biomedical text mining incorporates ideas from natural language processing, bioinformatics, medical informatics and computational linguistics. The strategies in this field have been applied to the biomedical literature available through services such as PubMed.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.

Chromosome conformation capture techniques are a set of molecular biology methods used to analyze the spatial organization of chromatin in a cell. These methods quantify the number of interactions between genomic loci that are nearby in 3-D space, but may be separated by many nucleotides in the linear genome. Such interactions may result from biological functions, such as promoter-enhancer interactions, or from random polymer looping, where undirected physical motion of chromatin causes loci to collide. Interaction frequencies may be analyzed directly, or they may be converted to distances and used to reconstruct 3-D structures.

Jason H. Moore is a translational bioinformatics scientist, biomedical informatician, and human geneticist, the Edward Rose Professor of Informatics and Director of the Institute for Biomedical Informatics at the Perelman School of Medicine at the University of Pennsylvania, where he is also Senior Associate Dean for Informatics and Director of the Division of Informatics in the Department of Biostatistics, Epidemiology, and Informatics.
Relief is an algorithm developed by Kira and Rendell in 1992 that takes a filter-method approach to feature selection that is notably sensitive to feature interactions. It was originally designed for application to binary classification problems with discrete or numerical features. Relief calculates a feature score for each feature which can then be applied to rank and select top scoring features for feature selection. Alternatively, these scores may be applied as feature weights to guide downstream modeling. Relief feature scoring is based on the identification of feature value differences between nearest neighbor instance pairs. If a feature value difference is observed in a neighboring instance pair with the same class, the feature score decreases. Alternatively, if a feature value difference is observed in a neighboring instance pair with different class values, the feature score increases. The original Relief algorithm has since inspired a family of Relief-based feature selection algorithms (RBAs), including the ReliefF algorithm. Beyond the original Relief algorithm, RBAs have been adapted to (1) perform more reliably in noisy problems, (2) generalize to multi-class problems (3) generalize to numerical outcome problems, and (4) to make them robust to incomplete data.
Flow cytometry bioinformatics is the application of bioinformatics to flow cytometry data, which involves storing, retrieving, organizing and analyzing flow cytometry data using extensive computational resources and tools. Flow cytometry bioinformatics requires extensive use of and contributes to the development of techniques from computational statistics and machine learning. Flow cytometry and related methods allow the quantification of multiple independent biomarkers on large numbers of single cells. The rapid growth in the multidimensionality and throughput of flow cytometry data, particularly in the 2000s, has led to the creation of a variety of computational analysis methods, data standards, and public databases for the sharing of results.
Genetic reductionism is the belief that understanding genes is sufficient to understand all aspects of human behavior. It is a specific form of reductionism and of biological determinism, based on a perspective which defines genes as distinct units of information with consistent properties. It also covers attempts to define specific phenomena in exclusively genetic terms, as in the case of the "warrior gene".
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.

Epistasis is a phenomenon in genetics in which the effect of a gene mutation is dependent on the presence or absence of mutations in one or more other genes, respectively termed modifier genes. In other words, the effect of the mutation is dependent on the genetic background in which it appears. Epistatic mutations therefore have different effects on their own than when they occur together. Originally, the term epistasis specifically meant that the effect of a gene variant is masked by that of different gene.
Perturb-seq refers to a high-throughput method of performing single cell RNA sequencing (scRNA-seq) on pooled genetic perturbation screens. Perturb-seq combines multiplexed CRISPR mediated gene inactivations with single cell RNA sequencing to assess comprehensive gene expression phenotypes for each perturbation. Inferring a gene’s function by applying genetic perturbations to knock down or knock out a gene and studying the resulting phenotype is known as reverse genetics. Perturb-seq is a reverse genetics approach that allows for the investigation of phenotypes at the level of the transcriptome, to elucidate gene functions in many cells, in a massively parallel fashion.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
References
- ↑ McKinney, Brett A.; Reif, David M.; Ritchie, Marylyn D.; Moore, Jason H. (1 January 2006). "Machine learning for detecting gene-gene interactions: a review". Applied Bioinformatics. 5 (2): 77–88. doi:10.2165/00822942-200605020-00002. ISSN 1175-5636. PMC 3244050 . PMID 16722772.
- ↑ Ritchie, Marylyn D.; Hahn, Lance W.; Roodi, Nady; Bailey, L. Renee; Dupont, William D.; Parl, Fritz F.; Moore, Jason H. (1 July 2001). "Multifactor-Dimensionality Reduction Reveals High-Order Interactions among Estrogen-Metabolism Genes in Sporadic Breast Cancer". The American Journal of Human Genetics. 69 (1): 138–147. doi:10.1086/321276. ISSN 0002-9297. PMC 1226028 . PMID 11404819.
- ↑ Ritchie, Marylyn D.; Hahn, Lance W.; Moore, Jason H. (1 February 2003). "Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, missing data, phenocopy, and genetic heterogeneity". Genetic Epidemiology. 24 (2): 150–157. doi:10.1002/gepi.10218. ISSN 1098-2272. PMID 12548676. S2CID 6335612.
- ↑ Hahn, L. W.; Ritchie, M. D.; Moore, J. H. (12 February 2003). "Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions". Bioinformatics. 19 (3): 376–382. doi: 10.1093/bioinformatics/btf869 . ISSN 1367-4803. PMID 12584123.
- ↑ W., Hahn, Lance; H., Moore, Jason (1 January 2004). "Ideal Discrimination of Discrete Clinical Endpoints Using Multilocus Genotypes". In Silico Biology. 4 (2): 183–194. ISSN 1386-6338. PMID 15107022.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Moore, Jason H. (1 November 2004). "Computational analysis of gene-gene interactions using multifactor dimensionality reduction". Expert Review of Molecular Diagnostics. 4 (6): 795–803. doi:10.1586/14737159.4.6.795. ISSN 1473-7159. PMID 15525222. S2CID 26324399.
- 1 2 Moore, JasonH.; Andrews, PeterC. (1 January 2015). "Epistasis Analysis Using Multifactor Dimensionality Reduction". In Moore, Jason H.; Williams, Scott M. (eds.). Epistasis. Methods in Molecular Biology. Vol. 1253. Springer New York. pp. 301–314. doi:10.1007/978-1-4939-2155-3_16. ISBN 9781493921546. PMID 25403539.
- ↑ Moore, Jason H. (1 January 2010). "Detecting, characterizing, and interpreting nonlinear gene-gene interactions using multifactor dimensionality reduction". Advances in Genetics. 72: 101–116. doi:10.1016/B978-0-12-380862-2.00005-9. ISBN 978-0-12-380862-2. ISSN 0065-2660. PMID 21029850.
- 1 2 Moore, Jason H.; Gilbert, Joshua C.; Tsai, Chia-Ti; Chiang, Fu-Tien; Holden, Todd; Barney, Nate; White, Bill C. (21 July 2006). "A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility". Journal of Theoretical Biology. 241 (2): 252–261. doi:10.1016/j.jtbi.2005.11.036. PMID 16457852.
- ↑ Michalski, R (February 1983). "A theory and methodology of inductive learning". Artificial Intelligence. 20 (2): 111–161. doi:10.1016/0004-3702(83)90016-4.
- ↑ Velez, Digna R.; White, Bill C.; Motsinger, Alison A.; Bush, William S.; Ritchie, Marylyn D.; Williams, Scott M.; Moore, Jason H. (1 May 2007). "A balanced accuracy function for epistasis modeling in imbalanced datasets using multifactor dimensionality reduction". Genetic Epidemiology. 31 (4): 306–315. doi:10.1002/gepi.20211. ISSN 0741-0395. PMID 17323372. S2CID 28156181.
- ↑ Namkung, Junghyun; Kim, Kyunga; Yi, Sungon; Chung, Wonil; Kwon, Min-Seok; Park, Taesung (1 February 2009). "New evaluation measures for multifactor dimensionality reduction classifiers in gene-gene interaction analysis". Bioinformatics. 25 (3): 338–345. doi: 10.1093/bioinformatics/btn629 . ISSN 1367-4811. PMID 19164302.
- ↑ Bush, William S.; Edwards, Todd L.; Dudek, Scott M.; McKinney, Brett A.; Ritchie, Marylyn D. (1 January 2008). "Alternative contingency table measures improve the power and detection of multifactor dimensionality reduction". BMC Bioinformatics. 9: 238. doi: 10.1186/1471-2105-9-238 . ISSN 1471-2105. PMC 2412877 . PMID 18485205.
- 1 2 Coffey, Christopher S.; Hebert, Patricia R.; Ritchie, Marylyn D.; Krumholz, Harlan M.; Gaziano, J. Michael; Ridker, Paul M.; Brown, Nancy J.; Vaughan, Douglas E.; Moore, Jason H. (1 January 2004). "An application of conditional logistic regression and multifactor dimensionality reduction for detecting gene-gene Interactions on risk of myocardial infarction: The importance of model validation". BMC Bioinformatics. 5: 49. doi: 10.1186/1471-2105-5-49 . ISSN 1471-2105. PMC 419697 . PMID 15119966.
- ↑ Motsinger, Alison A.; Ritchie, Marylyn D. (1 September 2006). "The effect of reduction in cross-validation intervals on the performance of multifactor dimensionality reduction". Genetic Epidemiology. 30 (6): 546–555. doi:10.1002/gepi.20166. ISSN 1098-2272. PMID 16800004. S2CID 20573232.
- ↑ Gory, Jeffrey J.; Sweeney, Holly C.; Reif, David M.; Motsinger-Reif, Alison A. (5 November 2012). "A comparison of internal model validation methods for multifactor dimensionality reduction in the case of genetic heterogeneity". BMC Research Notes. 5: 623. doi: 10.1186/1756-0500-5-623 . ISSN 1756-0500. PMC 3599301 . PMID 23126544.
- ↑ Winham, Stacey J.; Slater, Andrew J.; Motsinger-Reif, Alison A. (22 July 2010). "A comparison of internal validation techniques for multifactor dimensionality reduction". BMC Bioinformatics. 11: 394. doi: 10.1186/1471-2105-11-394 . ISSN 1471-2105. PMC 2920275 . PMID 20650002.
- ↑ Pattin, Kristine A.; White, Bill C.; Barney, Nate; Gui, Jiang; Nelson, Heather H.; Kelsey, Karl T.; Andrew, Angeline S.; Karagas, Margaret R.; Moore, Jason H. (1 January 2009). "A computationally efficient hypothesis testing method for epistasis analysis using multifactor dimensionality reduction". Genetic Epidemiology. 33 (1): 87–94. doi:10.1002/gepi.20360. ISSN 1098-2272. PMC 2700860 . PMID 18671250.
- ↑ Greene, Casey S.; Himmelstein, Daniel S.; Nelson, Heather H.; Kelsey, Karl T.; Williams, Scott M.; Andrew, Angeline S.; Karagas, Margaret R.; Moore, Jason H. (1 October 2009). "Enabling personal genomics with an explicit test of epistasis". Biocomputing 2010: Pacific Symposium on Biocomputing. World Scientific. pp. 327–336. doi:10.1142/9789814295291_0035. ISBN 9789814299473. PMC 2916690 . PMID 19908385.
- ↑ Dai, Hongying; Bhandary, Madhusudan; Becker, Mara; Leeder, J. Steven; Gaedigk, Roger; Motsinger-Reif, Alison A. (22 May 2012). "Global tests of P-values for multifactor dimensionality reduction models in selection of optimal number of target genes". BioData Mining. 5 (1): 3. doi: 10.1186/1756-0381-5-3 . ISSN 1756-0381. PMC 3508622 . PMID 22616673.
- ↑ Motsinger-Reif, Alison A. (30 December 2008). "The effect of alternative permutation testing strategies on the performance of multifactor dimensionality reduction". BMC Research Notes. 1: 139. doi: 10.1186/1756-0500-1-139 . ISSN 1756-0500. PMC 2631601 . PMID 19116021.
- ↑ Greene, Casey S.; Penrod, Nadia M.; Williams, Scott M.; Moore, Jason H. (2 June 2009). "Failure to Replicate a Genetic Association May Provide Important Clues About Genetic Architecture". PLOS ONE. 4 (6): e5639. Bibcode:2009PLoSO...4.5639G. doi: 10.1371/journal.pone.0005639 . ISSN 1932-6203. PMC 2685469 . PMID 19503614.
- ↑ Piette, Elizabeth R.; Moore, Jason H. (19 April 2017). "Improving the Reproducibility of Genetic Association Results Using Genotype Resampling Methods". Applications of Evolutionary Computation. Lecture Notes in Computer Science. Vol. 10199. pp. 96–108. doi:10.1007/978-3-319-55849-3_7. ISBN 978-3-319-55848-6.
- ↑ Moore, Jason H.; Hu, Ting (1 January 2015). "Epistasis Analysis Using Information Theory". Epistasis. Methods in Molecular Biology. Vol. 1253. pp. 257–268. doi:10.1007/978-1-4939-2155-3_13. ISBN 978-1-4939-2154-6. ISSN 1940-6029. PMID 25403536.
- ↑ Kim, Nora Chung; Andrews, Peter C.; Asselbergs, Folkert W.; Frost, H. Robert; Williams, Scott M.; Harris, Brent T.; Read, Cynthia; Askland, Kathleen D.; Moore, Jason H. (28 July 2012). "Gene ontology analysis of pairwise genetic associations in two genome-wide studies of sporadic ALS". BioData Mining. 5 (1): 9. doi: 10.1186/1756-0381-5-9 . ISSN 1756-0381. PMC 3463436 . PMID 22839596.
- ↑ Cheng, Samantha; Andrew, Angeline S.; Andrews, Peter C.; Moore, Jason H. (1 January 2016). "Complex systems analysis of bladder cancer susceptibility reveals a role for decarboxylase activity in two genome-wide association studies". BioData Mining. 9: 40. doi: 10.1186/s13040-016-0119-z . PMC 5154053 . PMID 27999618.
- ↑ Gola, Damian; Mahachie John, Jestinah M.; van Steen, Kristel; König, Inke R. (1 March 2016). "A roadmap to multifactor dimensionality reduction methods". Briefings in Bioinformatics. 17 (2): 293–308. doi:10.1093/bib/bbv038. ISSN 1477-4054. PMC 4793893 . PMID 26108231.
- ↑ Martin, E. R.; Ritchie, M. D.; Hahn, L.; Kang, S.; Moore, J. H. (1 February 2006). "A novel method to identify gene-gene effects in nuclear families: the MDR-PDT". Genetic Epidemiology. 30 (2): 111–123. doi:10.1002/gepi.20128. ISSN 0741-0395. PMID 16374833. S2CID 25772215.
- ↑ Lou, Xiang-Yang; Chen, Guo-Bo; Yan, Lei; Ma, Jennie Z.; Mangold, Jamie E.; Zhu, Jun; Elston, Robert C.; Li, Ming D. (1 October 2008). "A combinatorial approach to detecting gene-gene and gene-environment interactions in family studies". American Journal of Human Genetics. 83 (4): 457–467. doi:10.1016/j.ajhg.2008.09.001. ISSN 1537-6605. PMC 2561932 . PMID 18834969.
- ↑ Cattaert, Tom; Urrea, Víctor; Naj, Adam C.; De Lobel, Lizzy; De Wit, Vanessa; Fu, Mao; Mahachie John, Jestinah M.; Shen, Haiqing; Calle, M. Luz (22 April 2010). "FAM-MDR: a flexible family-based multifactor dimensionality reduction technique to detect epistasis using related individuals". PLOS ONE. 5 (4): e10304. Bibcode:2010PLoSO...510304C. doi: 10.1371/journal.pone.0010304 . ISSN 1932-6203. PMC 2858665 . PMID 20421984.
- ↑ Leem, Sangseob; Park, Taesung (14 March 2017). "An empirical fuzzy multifactor dimensionality reduction method for detecting gene-gene interactions". BMC Genomics. 18 (Suppl 2): 115. doi: 10.1186/s12864-017-3496-x . ISSN 1471-2164. PMC 5374597 . PMID 28361694.
- ↑ Gui, Jiang; Andrew, Angeline S.; Andrews, Peter; Nelson, Heather M.; Kelsey, Karl T.; Karagas, Margaret R.; Moore, Jason H. (1 January 2010). "A simple and computationally efficient sampling approach to covariate adjustment for multifactor dimensionality reduction analysis of epistasis". Human Heredity. 70 (3): 219–225. doi:10.1159/000319175. ISSN 1423-0062. PMC 2982850 . PMID 20924193.
- ↑ Chung, Yujin; Lee, Seung Yeoun; Elston, Robert C.; Park, Taesung (1 January 2007). "Odds ratio based multifactor-dimensionality reduction method for detecting gene-gene interactions". Bioinformatics. 23 (1): 71–76. doi: 10.1093/bioinformatics/btl557 . ISSN 1367-4811. PMID 17092990.
- ↑ Dai, Hongying; Charnigo, Richard J.; Becker, Mara L.; Leeder, J. Steven; Motsinger-Reif, Alison A. (8 January 2013). "Risk score modeling of multiple gene to gene interactions using aggregated-multifactor dimensionality reduction". BioData Mining. 6 (1): 1. doi: 10.1186/1756-0381-6-1 . PMC 3560267 . PMID 23294634.
- ↑ Gui, Jiang; Moore, Jason H.; Kelsey, Karl T.; Marsit, Carmen J.; Karagas, Margaret R.; Andrew, Angeline S. (1 January 2011). "A novel survival multifactor dimensionality reduction method for detecting gene-gene interactions with application to bladder cancer prognosis". Human Genetics. 129 (1): 101–110. doi:10.1007/s00439-010-0905-5. ISSN 1432-1203. PMC 3255326 . PMID 20981448.
- ↑ Lee, Seungyeoun; Son, Donghee; Yu, Wenbao; Park, Taesung (1 December 2016). "Gene-Gene Interaction Analysis for the Accelerated Failure Time Model Using a Unified Model-Based Multifactor Dimensionality Reduction Method". Genomics & Informatics. 14 (4): 166–172. doi:10.5808/GI.2016.14.4.166. ISSN 1598-866X. PMC 5287120 . PMID 28154507.
- ↑ Gui, Jiang; Andrew, Angeline S.; Andrews, Peter; Nelson, Heather M.; Kelsey, Karl T.; Karagas, Margaret R.; Moore, Jason H. (1 January 2011). "A robust multifactor dimensionality reduction method for detecting gene-gene interactions with application to the genetic analysis of bladder cancer susceptibility". Annals of Human Genetics. 75 (1): 20–28. doi:10.1111/j.1469-1809.2010.00624.x. ISSN 1469-1809. PMC 3057873 . PMID 21091664.
- ↑ Gui, Jiang; Moore, Jason H.; Williams, Scott M.; Andrews, Peter; Hillege, Hans L.; van der Harst, Pim; Navis, Gerjan; Van Gilst, Wiek H.; Asselbergs, Folkert W. (1 January 2013). "A Simple and Computationally Efficient Approach to Multifactor Dimensionality Reduction Analysis of Gene-Gene Interactions for Quantitative Traits". PLOS ONE. 8 (6): e66545. Bibcode:2013PLoSO...866545G. doi: 10.1371/journal.pone.0066545 . ISSN 1932-6203. PMC 3689797 . PMID 23805232.
- ↑ Lou, Xiang-Yang; Chen, Guo-Bo; Yan, Lei; Ma, Jennie Z.; Zhu, Jun; Elston, Robert C.; Li, Ming D. (1 June 2007). "A generalized combinatorial approach for detecting gene-by-gene and gene-by-environment interactions with application to nicotine dependence". American Journal of Human Genetics. 80 (6): 1125–1137. doi:10.1086/518312. ISSN 0002-9297. PMC 1867100 . PMID 17503330.
- ↑ Tsai, Chia-Ti; Lai, Ling-Ping; Lin, Jiunn-Lee; Chiang, Fu-Tien; Hwang, Juey-Jen; Ritchie, Marylyn D.; Moore, Jason H.; Hsu, Kuan-Lih; Tseng, Chuen-Den (6 April 2004). "Renin-Angiotensin System Gene Polymorphisms and Atrial Fibrillation". Circulation. 109 (13): 1640–1646. doi: 10.1161/01.CIR.0000124487.36586.26 . ISSN 0009-7322. PMID 15023884.
- ↑ Asselbergs, Folkert W.; Moore, Jason H.; van den Berg, Maarten P.; Rimm, Eric B.; de Boer, Rudolf A.; Dullaart, Robin P.; Navis, Gerjan; van Gilst, Wiek H. (1 January 2006). "A role for CETP TaqIB polymorphism in determining susceptibility to atrial fibrillation: a nested case control study". BMC Medical Genetics. 7: 39. doi: 10.1186/1471-2350-7-39 . ISSN 1471-2350. PMC 1462991 . PMID 16623947.
- ↑ Ma, D.Q.; Whitehead, P.L.; Menold, M.M.; Martin, E.R.; Ashley-Koch, A.E.; Mei, H.; Ritchie, M.D.; DeLong, G.R.; Abramson, R.K. (1 September 2005). "Identification of Significant Association and Gene-Gene Interaction of GABA Receptor Subunit Genes in Autism". The American Journal of Human Genetics. 77 (3): 377–388. doi:10.1086/433195. ISSN 0002-9297. PMC 1226204 . PMID 16080114.
- ↑ Andrew, Angeline S.; Nelson, Heather H.; Kelsey, Karl T.; Moore, Jason H.; Meng, Alexis C.; Casella, Daniel P.; Tosteson, Tor D.; Schned, Alan R.; Karagas, Margaret R. (1 May 2006). "Concordance of multiple analytical approaches demonstrates a complex relationship between DNA repair gene SNPs, smoking and bladder cancer susceptibility". Carcinogenesis. 27 (5): 1030–1037. doi: 10.1093/carcin/bgi284 . ISSN 0143-3334. PMID 16311243.
- ↑ Andrew, Angeline S.; Karagas, Margaret R.; Nelson, Heather H.; Guarrera, Simonetta; Polidoro, Silvia; Gamberini, Sara; Sacerdote, Carlotta; Moore, Jason H.; Kelsey, Karl T. (1 January 2008). "DNA Repair Polymorphisms Modify Bladder Cancer Risk: A Multi-factor Analytic Strategy". Human Heredity. 65 (2): 105–118. doi:10.1159/000108942. ISSN 0001-5652. PMC 2857629 . PMID 17898541.
- ↑ Andrew, Angeline S.; Hu, Ting; Gu, Jian; Gui, Jiang; Ye, Yuanqing; Marsit, Carmen J.; Kelsey, Karl T.; Schned, Alan R.; Tanyos, Sam A. (1 January 2012). "HSD3B and gene-gene interactions in a pathway-based analysis of genetic susceptibility to bladder cancer". PLOS ONE. 7 (12): e51301. Bibcode:2012PLoSO...751301A. doi: 10.1371/journal.pone.0051301 . ISSN 1932-6203. PMC 3526593 . PMID 23284679.
- ↑ Cao, Jingjing; Luo, Chenglin; Yan, Rui; Peng, Rui; Wang, Kaijuan; Wang, Peng; Ye, Hua; Song, Chunhua (1 December 2016). "rs15869 at miRNA binding site in BRCA2 is associated with breast cancer susceptibility". Medical Oncology. 33 (12): 135. doi:10.1007/s12032-016-0849-2. ISSN 1357-0560. PMID 27807724. S2CID 26042128.
- ↑ Williams, Scott M.; Ritchie, Marylyn D.; III, John A. Phillips; Dawson, Elliot; Prince, Melissa; Dzhura, Elvira; Willis, Alecia; Semenya, Amma; Summar, Marshall (1 January 2004). "Multilocus Analysis of Hypertension: A Hierarchical Approach". Human Heredity. 57 (1): 28–38. doi:10.1159/000077387. ISSN 0001-5652. PMID 15133310. S2CID 21079485.
- ↑ Sanada, Hironobu; Yatabe, Junichi; Midorikawa, Sanae; Hashimoto, Shigeatsu; Watanabe, Tsuyoshi; Moore, Jason H.; Ritchie, Marylyn D.; Williams, Scott M.; Pezzullo, John C. (1 March 2006). "Single-Nucleotide Polymorphisms for Diagnosis of Salt-Sensitive Hypertension". Clinical Chemistry. 52 (3): 352–360. doi: 10.1373/clinchem.2005.059139 . ISSN 0009-9147. PMID 16439609.
- ↑ Moore, Jason H.; Williams, Scott M. (1 January 2002). "New strategies for identifying gene-gene interactions in hypertension". Annals of Medicine. 34 (2): 88–95. doi:10.1080/07853890252953473. ISSN 0785-3890. PMID 12108579. S2CID 25398042.
- ↑ De, Rishika; Verma, Shefali S.; Holzinger, Emily; Hall, Molly; Burt, Amber; Carrell, David S.; Crosslin, David R.; Jarvik, Gail P.; Kuivaniemi, Helena (1 February 2017). "Identifying gene-gene interactions that are highly associated with four quantitative lipid traits across multiple cohorts" (PDF). Human Genetics. 136 (2): 165–178. doi:10.1007/s00439-016-1738-7. ISSN 1432-1203. PMID 27848076. S2CID 24702049.
- ↑ De, Rishika; Verma, Shefali S.; Drenos, Fotios; Holzinger, Emily R.; Holmes, Michael V.; Hall, Molly A.; Crosslin, David R.; Carrell, David S.; Hakonarson, Hakon (1 January 2015). "Identifying gene-gene interactions that are highly associated with Body Mass Index using Quantitative Multifactor Dimensionality Reduction (QMDR)". BioData Mining. 8: 41. doi: 10.1186/s13040-015-0074-0 . PMC 4678717 . PMID 26674805.
- ↑ Duell, Eric J.; Bracci, Paige M.; Moore, Jason H.; Burk, Robert D.; Kelsey, Karl T.; Holly, Elizabeth A. (1 June 2008). "Detecting pathway-based gene-gene and gene-environment interactions in pancreatic cancer". Cancer Epidemiology, Biomarkers & Prevention. 17 (6): 1470–1479. doi:10.1158/1055-9965.EPI-07-2797. ISSN 1055-9965. PMC 4410856 . PMID 18559563.
- ↑ Xu, Jianfeng; Lowey, James; Wiklund, Fredrik; Sun, Jielin; Lindmark, Fredrik; Hsu, Fang-Chi; Dimitrov, Latchezar; Chang, Baoli; Turner, Aubrey R. (1 November 2005). "The Interaction of Four Genes in the Inflammation Pathway Significantly Predicts Prostate Cancer Risk". Cancer Epidemiology, Biomarkers & Prevention. 14 (11): 2563–2568. doi: 10.1158/1055-9965.EPI-05-0356 . ISSN 1055-9965. PMID 16284379.
- ↑ Lavender, Nicole A.; Rogers, Erica N.; Yeyeodu, Susan; Rudd, James; Hu, Ting; Zhang, Jie; Brock, Guy N.; Kimbro, Kevin S.; Moore, Jason H. (30 April 2012). "Interaction among apoptosis-associated sequence variants and joint effects on aggressive prostate cancer". BMC Medical Genomics. 5: 11. doi: 10.1186/1755-8794-5-11 . ISSN 1755-8794. PMC 3355002 . PMID 22546513.
- ↑ Lavender, Nicole A.; Benford, Marnita L.; VanCleave, Tiva T.; Brock, Guy N.; Kittles, Rick A.; Moore, Jason H.; Hein, David W.; Kidd, La Creis R. (16 November 2009). "Examination of polymorphic glutathione S-transferase (GST) genes, tobacco smoking and prostate cancer risk among men of African descent: a case-control study". BMC Cancer. 9: 397. doi: 10.1186/1471-2407-9-397 . ISSN 1471-2407. PMC 2783040 . PMID 19917083.
- ↑ Collins, Ryan L.; Hu, Ting; Wejse, Christian; Sirugo, Giorgio; Williams, Scott M.; Moore, Jason H. (18 February 2013). "Multifactor dimensionality reduction reveals a three-locus epistatic interaction associated with susceptibility to pulmonary tuberculosis". BioData Mining. 6 (1): 4. doi: 10.1186/1756-0381-6-4 . PMC 3618340 . PMID 23418869.
- ↑ Wilke, Russell A.; Reif, David M.; Moore, Jason H. (1 November 2005). "Combinatorial Pharmacogenetics". Nature Reviews Drug Discovery. 4 (11): 911–918. doi:10.1038/nrd1874. ISSN 1474-1776. PMID 16264434. S2CID 11643026.
- ↑ Motsinger, Alison A.; Ritchie, Marylyn D.; Shafer, Robert W.; Robbins, Gregory K.; Morse, Gene D.; Labbe, Line; Wilkinson, Grant R.; Clifford, David B.; D'Aquila, Richard T. (1 November 2006). "Multilocus genetic interactions and response to efavirenz-containing regimens: an adult AIDS clinical trials group study". Pharmacogenetics and Genomics. 16 (11): 837–845. doi:10.1097/01.fpc.0000230413.97596.fa. ISSN 1744-6872. PMID 17047492. S2CID 26266170.
- ↑ Ritchie, Marylyn D.; Motsinger, Alison A. (1 December 2005). "Multifactor dimensionality reduction for detecting gene-gene and gene-environment interactions in pharmacogenomics studies". Pharmacogenomics. 6 (8): 823–834. doi:10.2217/14622416.6.8.823. ISSN 1462-2416. PMID 16296945. S2CID 10348021.
- ↑ Moore, Jason H.; Asselbergs, Folkert W.; Williams, Scott M. (15 February 2010). "Bioinformatics challenges for genome-wide association studies". Bioinformatics. 26 (4): 445–455. doi:10.1093/bioinformatics/btp713. ISSN 1367-4811. PMC 2820680 . PMID 20053841.
- ↑ Sun, Xiangqing; Lu, Qing; Mukherjee, Shubhabrata; Mukheerjee, Shubhabrata; Crane, Paul K.; Elston, Robert; Ritchie, Marylyn D. (1 January 2014). "Analysis pipeline for the epistasis search – statistical versus biological filtering". Frontiers in Genetics. 5: 106. doi: 10.3389/fgene.2014.00106 . PMC 4012196 . PMID 24817878.
- ↑ Pendergrass, Sarah A.; Frase, Alex; Wallace, John; Wolfe, Daniel; Katiyar, Neerja; Moore, Carrie; Ritchie, Marylyn D. (30 December 2013). "Genomic analyses with biofilter 2.0: knowledge driven filtering, annotation, and model development". BioData Mining. 6 (1): 25. doi: 10.1186/1756-0381-6-25 . PMC 3917600 . PMID 24378202.
- ↑ Moore, Jason H. (1 January 2015). "Epistasis Analysis Using ReliefF". Epistasis. Methods in Molecular Biology. Vol. 1253. pp. 315–325. doi:10.1007/978-1-4939-2155-3_17. ISBN 978-1-4939-2154-6. ISSN 1940-6029. PMID 25403540.
- ↑ Moore, Jason H.; White, Bill C. (1 January 2007). "Genome-Wide Genetic Analysis Using Genetic Programming: The Critical Need for Expert Knowledge". In Riolo, Rick; Soule, Terence; Worzel, Bill (eds.). Genetic Programming Theory and Practice IV. Genetic and Evolutionary Computation. Springer US. pp. 11–28. doi:10.1007/978-0-387-49650-4_2. ISBN 9780387333755. S2CID 55188394.
- ↑ Greene, Casey S.; Sinnott-Armstrong, Nicholas A.; Himmelstein, Daniel S.; Park, Paul J.; Moore, Jason H.; Harris, Brent T. (1 March 2010). "Multifactor dimensionality reduction for graphics processing units enables genome-wide testing of epistasis in sporadic ALS". Bioinformatics. 26 (5): 694–695. doi:10.1093/bioinformatics/btq009. ISSN 1367-4811. PMC 2828117 . PMID 20081222.
- ↑ Bush, William S.; Dudek, Scott M.; Ritchie, Marylyn D. (1 September 2006). "Parallel multifactor dimensionality reduction: a tool for the large-scale analysis of gene-gene interactions". Bioinformatics. 22 (17): 2173–2174. doi:10.1093/bioinformatics/btl347. ISSN 1367-4811. PMC 4939609 . PMID 16809395.
- ↑ Sinnott-Armstrong, Nicholas A.; Greene, Casey S.; Cancare, Fabio; Moore, Jason H. (24 July 2009). "Accelerating epistasis analysis in human genetics with consumer graphics hardware". BMC Research Notes. 2: 149. doi: 10.1186/1756-0500-2-149 . ISSN 1756-0500. PMC 2732631 . PMID 19630950.
- ↑ Winham, Stacey J.; Motsinger-Reif, Alison A. (16 August 2011). "An R package implementation of multifactor dimensionality reduction". BioData Mining. 4 (1): 24. doi: 10.1186/1756-0381-4-24 . ISSN 1756-0381. PMC 3177775 . PMID 21846375.
- ↑ Calle, M. Luz; Urrea, Víctor; Malats, Núria; Van Steen, Kristel (1 September 2010). "mbmdr: an R package for exploring gene-gene interactions associated with binary or quantitative traits". Bioinformatics. 26 (17): 2198–2199. doi: 10.1093/bioinformatics/btq352 . ISSN 1367-4811. PMID 20595460.