Quantum chemistry, also called molecular quantum mechanics, is a branch of chemistry focused on the application of quantum mechanics in physical models and experiments of chemical systems. Understanding electronic structure and molecular dynamics using the Schrödinger equations are central topics in quantum chemistry.

In chemistry and physics, activation energy is the energy that must be provided to compounds to result in a chemical reaction.The activation energy (Ea) of a reaction is measured in joules per mole (J/mol), kilojoules per mole (kJ/mol) or kilocalories per mole (kcal/mol). Activation energy can be thought of as the magnitude of the potential barrier separating minima of the potential energy surface pertaining to the initial and final thermodynamic state. For a chemical reaction to proceed at a reasonable rate, the temperature of the system should be high enough such that there exists an appreciable number of molecules with translational energy equal to or greater than the activation energy. The term Activation Energy was introduced in 1889 by the Swedish scientist Svante Arrhenius.

Roentgenium is a chemical element with the symbol Rg and atomic number 111. It is an extremely radioactive synthetic element that can be created in a laboratory but is not found in nature. The most stable known isotope, roentgenium-282, has a half-life of 100 seconds, although the unconfirmed roentgenium-286 may have a longer half-life of about 10.7 minutes. Roentgenium was first created in 1994 by the GSI Helmholtz Centre for Heavy Ion Research near Darmstadt, Germany. It is named after the physicist Wilhelm Röntgen, who discovered X-rays.
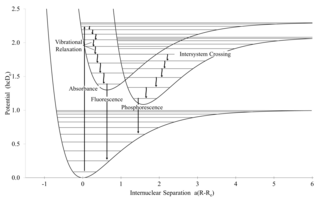
Intersystem crossing (ISC) is a radiationless process involving a transition between the two electronic states with different states spin multiplicity.
Dynamic nuclear polarization (DNP) results from transferring spin polarization from electrons to nuclei, thereby aligning the nuclear spins to the extent that electron spins are aligned. Note that the alignment of electron spins at a given magnetic field and temperature is described by the Boltzmann distribution under the thermal equilibrium. It is also possible that those electrons are aligned to a higher degree of order by other preparations of electron spin order such as: chemical reactions, optical pumping and spin injection. DNP is considered one of several techniques for hyperpolarization. DNP can also be induced using unpaired electrons produced by radiation damage in solids.
In physics and chemistry, a selection rule, or transition rule, formally constrains the possible transitions of a system from one quantum state to another. Selection rules have been derived for electromagnetic transitions in molecules, in atoms, in atomic nuclei, and so on. The selection rules may differ according to the technique used to observe the transition. The selection rule also plays a role in chemical reactions, where some are formally spin-forbidden reactions, that is, reactions where the spin state changes at least once from reactants to products.
Singlet oxygen, systematically named dioxygen(singlet) and dioxidene, is a gaseous inorganic chemical with the formula O=O, which is in a quantum state where all electrons are spin paired. It is kinetically unstable at ambient temperature, however the rate of decay is slow.
Vibronic coupling in a molecule involves the interaction between electronic and nuclear vibrational motion. The term "vibronic" originates from the combination of the terms "vibrational" and "electronic", denoting the idea that in a molecule, vibrational and electronic interactions are interrelated and influence each other. The magnitude of vibronic coupling reflects the degree of such interrelation.
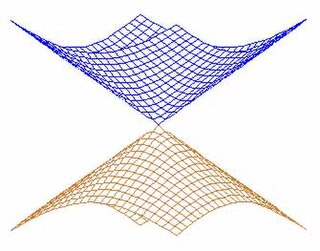
In quantum chemistry, a conical intersection of two or more potential energy surfaces is the set of molecular geometry points where the potential energy surfaces are degenerate (intersect) and the non-adiabatic couplings between these states are non-vanishing. In the vicinity of conical intersections, the Born–Oppenheimer approximation breaks down and the coupling between electronic and nuclear motion becomes important, allowing non-adiabatic processes to take place. The location and characterization of conical intersections are therefore essential to the understanding of a wide range of important phenomena governed by non-adiabatic events, such as photoisomerization, photosynthesis, vision and the photostability of DNA. The conical intersection involving the ground electronic state potential energy surface of the C6H3F3+ molecular ion is discussed in connection with the Jahn-Teller effect in Section 13.4.2 on pages 380-388 of the textbook by Bunker and Jensen.
Marcus theory is a theory originally developed by Rudolph A. Marcus, starting in 1956, to explain the rates of electron transfer reactions – the rate at which an electron can move or jump from one chemical species (called the electron donor) to another (called the electron acceptor). It was originally formulated to address outer sphere electron transfer reactions, in which the two chemical species only change in their charge with an electron jumping (e.g. the oxidation of an ion like Fe2+/Fe3+), but do not undergo large structural changes. It was extended to include inner sphere electron transfer contributions, in which a change of distances or geometry in the solvation or coordination shells of the two chemical species is taken into account (the Fe-O distances in Fe(H2O)2+ and Fe(H2O)3+ are different).

Transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.

The Landau–Zener formula is an analytic solution to the equations of motion governing the transition dynamics of a two-state quantum system, with a time-dependent Hamiltonian varying such that the energy separation of the two states is a linear function of time. The formula, giving the probability of a diabatic transition between the two energy states, was published separately by Lev Landau, Clarence Zener, Ernst Stueckelberg, and Ettore Majorana, in 1932.
Xenon monochloride (XeCl) is an exciplex which is used in excimer lasers and excimer lamps emitting near ultraviolet light at 308 nm. It is most commonly used in medicine. Xenon monochloride was first synthesized in the 1960s. Its kinetic scheme is very complex and its state changes occur on a nanosecond timescale. In the gaseous state, at least two kinds of xenon monochloride are known: XeCl and Xe
2Cl, whereas complex aggregates form in the solid state in noble gas matrices. The excited state of xenon resembles halogens and it reacts with them to form excited molecular compounds.

James Bernhart Anderson is Evan Pugh Professor of Chemistry and Physics at the Pennsylvania State University. He specializes in Quantum Chemistry by Monte Carlo methods, molecular dynamics of reactive collisions, kinetics and mechanisms of gas phase reactions, and rare-event theory.
Surface hopping is a mixed quantum-classical technique that incorporates quantum mechanical effects into molecular dynamics simulations. Traditional molecular dynamics assume the Born-Oppenheimer approximation, where the lighter electrons adjust instantaneously to the motion of the nuclei. Though the Born-Oppenheimer approximation is applicable to a wide range of problems, there are several applications, such as photoexcited dynamics, electron transfer, and surface chemistry where this approximation falls apart. Surface hopping partially incorporates the non-adiabatic effects by including excited adiabatic surfaces in the calculations, and allowing for 'hops' between these surfaces, subject to certain criteria.
Millard Henry Alexander is a Distinguished University Professor at the University of Maryland, with appointments in the Department of Chemistry and Biochemistry and the Institute for Physical Science and Technology. He is the author of over 300 publications and an active researcher in the fields of molecular collision dynamics and theoretical chemistry.
In chemistry, the selection rule formally restrict certain reactions, known as spin-forbidden reactions, from occurring due to a required change between two differing quantum states. When a reactant exists in one spin state and the product exists in a different spin state, the corresponding reaction will have an increased activation energy when compared to a similar reaction in which the spin states of the reactant and product are isomorphic. As a result of this increased activation energy, a decreased rate of reaction is observed.
In chemical kinetics, the Aquilanti–Mundim deformed Arrhenius model is a generalization of the standard Arrhenius law.
Semiclassical Transition State Theory (SCTST) is an efficient chemical rate theory, which aims to calculate accurate rate constants of chemical reactions, including nuclear quantum effects such as tunnelling, from ab initio quantum chemistry. The method makes use of the semiclassical WKB wavefunction, Bohr-sommerfeld theory and vibrational perturbation theory to derive an analytical relation for the probability of a particle transmitting through a potential barrier at some energy, E. It was first developed by Bill Miller and coworkers in the 1970's, and has been further developed to allow for application to larger systems and using more accurate potentials.
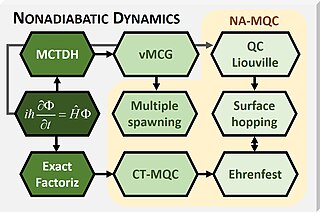
Mixed quantum-classical (MQC) dynamics is a class of computational theoretical chemistry methods tailored to simulate nonadiabatic (NA) processes in molecular and supramolecular chemistry. Such methods are characterized by:
- Propagation of nuclear dynamics through classical trajectories;
- Propagation of the electrons through quantum methods;
- A feedback algorithm between the electronic and nuclear subsystems to recover nonadiabatic information.