Diagnosis
| | This section is empty. You can help by adding to it. (March 2019) |
| Oculomotor apraxia | |
|---|---|
| Other names | Cogan ocular motor apraxia or saccadic initiation failure |
| Specialty | Ophthalmology |
Oculomotor apraxia (OMA) is the absence or defect of controlled, voluntary, and purposeful eye movement. [1] It was first described in 1952 by the American ophthalmologist David Glendenning Cogan. [2] People with this condition have difficulty moving their eyes horizontally and moving them quickly. The main difficulty is in saccade initiation, but there is also impaired cancellation of the vestibulo-ocular reflex. Patients have to turn their head in order to compensate for the lack of eye movement initiation in order to follow an object or see objects in their peripheral vision, but they often exceed their target. There is controversy regarding whether OMA should be considered an apraxia, since apraxia is the inability to perform a learned or skilled motor action to command, and saccade initiation is neither a learned nor a skilled action. [3]
Even though OMA is not always associated with developmental issues, children with this condition often have hypotonia, decreased muscle tone, and show developmental delays. Some common delays are seen in speech, reading and motor development. [3]
OMA is a neurological condition. Although some brain imaging studies of people with OMA reveal a normal brain, some MRI studies have revealed unusual appearance of some brain areas, in particular the corpus callosum, cerebellum, and fourth ventricle. Oculomotor apraxia can be acquired or congenital. Sometimes no cause is found, in which case it is described as idiopathic. [1]
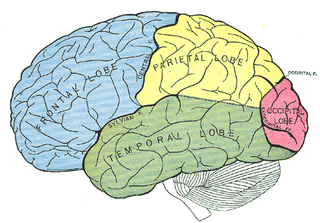
A person may be born with the parts of the brain for eye movement control not working, or may manifest poor eye movement control in childhood. If any part of the brain that controls eye movement becomes damaged, then OMA may develop. [2] One of the potential causes is bifrontal hemorrhages. In this case, OMA is associated with bilateral lesions of the frontal eye fields (FEF), located in the caudal middle frontal gyrus. The FEF control voluntary eye movements, including saccades, smooth pursuit and vergence. OMA can also be associated with bilateral hemorrhages in the parietal eye fields (PEF). The PEF surround the posterior, medial segment of the intraparietal sulcus. They have a role in reflexive saccades, and send information to the FEF. Since the FEF and PEF have complementary roles in voluntary and reflexive production of saccades, respectively, and they get inputs from different areas of the brain, only bilateral lesions to both the FEF and PEF will cause persistent OMA. Patients with either bilateral FEF or bilateral PEF damage (but not both FEF and PEF) have been shown to regain at least some voluntary saccadic initiation some time after their hemorrhages. Other causes of OMA include brain tumors and cardiovascular problems. [4] [5]
A subgroup of genetically recessive ataxias associated with OMA has been identified, with an onset during childhood. These are ataxia with oculomotor apraxia type 1 (AOA1), ataxia with oculomotor apraxia 2 (AOA2), and ataxia telangiectasia. These are autosomal recessive disorders and the associated gene products are involved in DNA repair. Both horizontal and vertical eye movements are affected in these disorders. [3] Although people with either type may have some mild cognitive problems, such as difficulty with concentration or performing multi-step activities, intellectual function is usually not affected. [6]
Ataxia-oculomotor apraxia type 1 (AOA1) usually has an onset of symptoms during childhood. It is an autosomal recessive cerebellar ataxia (ARCA) associated with hypoalbuminemia and hypercholesterolemia. Mutations in the gene APTX, which encodes for aprataxin, have been identified to be responsible for AOA1. Elevated creatine kinase is occasionally present, in addition to a sensorimotor axonal neuropathy, as shown by nerve conduction velocity studies. In addition, MRI studies have shown cerebellar atrophy, mild brainstem atrophy, and, in advanced cases, cortical atrophy. [7]
The aprataxin protein APTX can remove obstructive termini from DNA strand breaks that interfere with DNA repair. [8] APTX is recruited to DNA single-strand breaks by XRCC1 protein, where it functions as a nick sensor to scan the single-strand breaks for 5'-AMP obstructive termini that are intermediates in failed DNA ligase reactions. The removal of these obstructions allows DNA repair of the break to be completed. It is not yet clear which specific single-strand breaks are the neurodegenerative agents in AOA1 patients that lack functional aprataxin protein. However, single-strand breaks with 5'-AMP termini appear to be the most likely candidate lesion. [8]
Ataxia-oculomotor apraxia type 2 (AOA2), also known as spinocerebellar ataxia with axonal neuropathy type 2, [9] has its onset during adolescence. It is characterized by cerebellar atrophy and peripheral neuropathy. Sufferers of type 2 have high amounts of another protein called alpha-fetoprotein (AFP), and may also have high amounts of the protein creatine phosphokinase (CPK). Mutations in the SETX gene are the cause of the disease. AOA2 shows cerebellar atrophy, loss of Purkinje cells, and demyelination. In particular, there is a failure of the cerebrocerebellar circuit in AOA2 that has been shown to be responsible for the weaker coordination of complex cognitive functions such as working memory, executive functions, speech, and sequence learning. [6] Although there is no sign of mental retardation or severe dementia, even after long disease duration, research on families with possible AOA2 have shown mild cognitive impairment as indexed by the mini–mental state examination (MMSE) and the Mattis dementia rating scale. These impairments appear to be mostly due to a deficit in initiation and concept subtests. [10] [11]
Telangiectasias are widened blood vessels that can develop anywhere on the skin, mucous membranes, whites of the eyes, and even in the brain. Telangiectasias are associated with multiple systemic signs, the most serious of which are unusual sensitivity to ionizing radiation, excessive chromosomal breakage, and a deficiency in the immune system. Ataxia telangiectasia results from defects in the ataxia telangiectasia mutated gene, which can cause abnormal cell death in various places of the body, including brain areas related to coordinated movement of the eyes. Patients with ataxia telangiectasia have prolonged vertical and horizontal saccade latencies and hypometric saccades, and, although not all, some patients show head thrusts. [3] [10] [12]
| | This section is empty. You can help by adding to it. (March 2019) |
Ataxia is a neurological sign consisting of lack of voluntary coordination of muscle movements that can include gait abnormality, speech changes, and abnormalities in eye movements, that indicates dysfunction of parts of the nervous system that coordinate movement, such as the cerebellum.
Ataxia–telangiectasia, also referred to as ataxia–telangiectasia syndrome or Louis–Bar syndrome, is a rare, neurodegenerative disease causing severe disability. Ataxia refers to poor coordination and telangiectasia to small dilated blood vessels, both of which are hallmarks of the disease. A–T affects many parts of the body:
Bálint's syndrome is an uncommon and incompletely understood triad of severe neuropsychological impairments: inability to perceive the visual field as a whole (simultanagnosia), difficulty in fixating the eyes, and inability to move the hand to a specific object by using vision. It was named in 1909 for the Austro-Hungarian neurologist and psychiatrist Rezső Bálint who first identified it.

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.
Dysmetria is a lack of coordination of movement typified by the undershoot or overshoot of intended position with the hand, arm, leg, or eye. It is a type of ataxia. It can also include an inability to judge distance or scale.
Sensory ataxia is both a symptom and a sign in neurology. It is a form of ataxia caused not by cerebellar dysfunction but by loss of sensory input into the control of movement.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.
Cerebellar ataxia is a form of ataxia originating in the cerebellum. Non-progressive congenital ataxia (NPCA) is a classical presentation of cerebral ataxias.
Dominant optic atrophy (DOA), or autosomal dominant optic atrophy (ADOA), (Kjer's type) is an autosomally inherited disease that affects the optic nerves, causing reduced visual acuity and blindness beginning in childhood. However, the disease can seem to re-present a second time with further vision loss due to the early onset of presbyopia symptoms (i.e., difficulty in viewing objects up close). DOA is characterized as affecting neurons called retinal ganglion cells (RGCs). This condition is due to mitochondrial dysfunction mediating the death of optic nerve fibers. The RGCs axons form the optic nerve. Therefore, the disease can be considered of the central nervous system. Dominant optic atrophy was first described clinically by Batten in 1896 and named Kjer’s optic neuropathy in 1959 after Danish ophthalmologist Poul Kjer, who studied 19 families with the disease. Although dominant optic atrophy is the most common autosomally inherited optic neuropathy (i.e., disease of the optic nerves), it is often misdiagnosed.

Supplementary eye field (SEF) is the name for the anatomical area of the dorsal medial frontal lobe of the primate cerebral cortex that is indirectly involved in the control of saccadic eye movements. Evidence for a supplementary eye field was first shown by Schlag, and Schlag-Rey. Current research strives to explore the SEF's contribution to visual search and its role in visual salience. The SEF constitutes together with the frontal eye fields (FEF), the intraparietal sulcus (IPS), and the superior colliculus (SC) one of the most important brain areas involved in the generation and control of eye movements, particularly in the direction contralateral to their location. Its precise function is not yet fully known. Neural recordings in the SEF show signals related to both vision and saccades somewhat like the frontal eye fields and superior colliculus, but currently most investigators think that the SEF has a special role in high level aspects of saccade control, like complex spatial transformations, learned transformations, and executive cognitive functions.

Aprataxin is a protein that in humans is encoded by the APTX gene.

Probable helicase senataxin is an enzyme that in humans is encoded by the SETX gene.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.

Mihai Ioan Botez was born in Ploiești, Romania, trained at Carol Davila University of Medicine and Pharmacy, a neurologist and academic who specialized in the field of neuropsychology. He immigrated to Montreal in the 1970s, becoming a professor at the Université de Montréal and director of the department of Neurology at the hospital Hôtel-Dieu de Montréal.
Harding ataxia is an autosomal recessive cerebellar ataxia originally described by Harding in 1981. This form of cerebellar ataxia is similar to Friedreich ataxia including that it results in poor reflexes and balance, but differs in several ways, including the absence of diabetes mellitus, optic atrophy, cardiomyopathy, skeletal abnormalities, and the fact that tendon reflexes in the arms and knees remain intact. This form of ataxia is characterized by onset in the first 20 years, and is less severe than Friedreich ataxia. Additional cases were diagnosed in 1989, 1990, 1991, and 1998.

Autosomal recessive cerebellar ataxia type 1 (ARCA1) is a condition characterized by progressive problems with movement. Signs and symptoms of the disorder first appear in early to mid-adulthood. People with this condition initially experience impaired speech (dysarthria), problems with coordination and balance (ataxia), or both. They may also have difficulty with movements that involve judging distance or scale (dysmetria). Other features of ARCA1 include abnormal eye movements (nystagmus) and problems following the movements of objects with their eyes. The movement problems are slowly progressive, often resulting in the need for a cane, walker, or wheelchair.
Mitohondrial optic neuropathies are a heterogenous group of disorders that present with visual disturbances resultant from mitochondrial dysfunction within the anatomy of the Retinal Ganglion Cells (RGC), optic nerve, optic chiasm, and optic tract. These disturbances are multifactorial, their aetiology consisting of metabolic and/or structural damage as a consequence of genetic mutations, environmental stressors, or both. The three most common neuro-ophthalmic abnormalities seen in mitochondrial disorders are bilateral optic neuropathy, ophthalmoplegia with ptosis, and pigmentary retinopathy.
Autosomal recessive cerebellar ataxia describes a heterogeneous group of rare genetic disorders with an autosomal recessive inheritance pattern and a clinical phenotype involving cerebellar ataxia.
Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS) is an autosomal recessive late-onset heredodegenerative multisystem neurological disease. The symptoms include poor balance and difficulty walking. Chronic cough and difficulty swallowing may also be present. Clinical findings include ataxia, sensory neuropathy, and absence of the vestibulo–ocular reflex. The syndrome was initially described in 2004. In 2019, the cause was identified as biallelic pentanucleotide expansion in the RFC1 gene.
Autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCADN) is a rare progressive genetic disorder that primarily affects the nervous system and is characterized by sensorineural hearing loss, narcolepsy with cataplexy, and dementia later in life. People with this disorder usually start showing symptoms when they are in their early-mid adulthoods. It is a type of autosomal dominant cerebellar ataxia.