Related Research Articles

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.
Archaeogenetics is the study of ancient DNA using various molecular genetic methods and DNA resources. This form of genetic analysis can be applied to human, animal, and plant specimens. Ancient DNA can be extracted from various fossilized specimens including bones, eggshells, and artificially preserved tissues in human and animal specimens. In plants, ancient DNA can be extracted from seeds and tissue. Archaeogenetics provides us with genetic evidence of ancient population group migrations, domestication events, and plant and animal evolution. The ancient DNA cross referenced with the DNA of relative modern genetic populations allows researchers to run comparison studies that provide a more complete analysis when ancient DNA is compromised.

AFLP-PCR or just AFLP is a PCR-based tool used in genetics research, DNA fingerprinting, and in the practice of genetic engineering. Developed in the early 1990s by KeyGene, AFLP uses restriction enzymes to digest genomic DNA, followed by ligation of adaptors to the sticky ends of the restriction fragments. A subset of the restriction fragments is then selected to be amplified. This selection is achieved by using primers complementary to the adaptor sequence, the restriction site sequence and a few nucleotides inside the restriction site fragments. The amplified fragments are separated and visualized on denaturing on agarose gel electrophoresis, either through autoradiography or fluorescence methodologies, or via automated capillary sequencing instruments.
Genotyping is the process of determining differences in the genetic make-up (genotype) of an individual by examining the individual's DNA sequence using biological assays and comparing it to another individual's sequence or a reference sequence. It reveals the alleles an individual has inherited from their parents. Traditionally genotyping is the use of DNA sequences to define biological populations by use of molecular tools. It does not usually involve defining the genes of an individual.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.

Combined Bisulfite Restriction Analysis is a molecular biology technique that allows for the sensitive quantification of DNA methylation levels at a specific genomic locus on a DNA sequence in a small sample of genomic DNA. The technique is a variation of bisulfite sequencing, and combines bisulfite conversion based polymerase chain reaction with restriction digestion. Originally developed to reliably handle minute amounts of genomic DNA from microdissected paraffin-embedded tissue samples, the technique has since seen widespread usage in cancer research and epigenetics studies.
Microfluidic whole genome haplotyping is a technique for the physical separation of individual chromosomes from a metaphase cell followed by direct resolution of the haplotype for each allele.
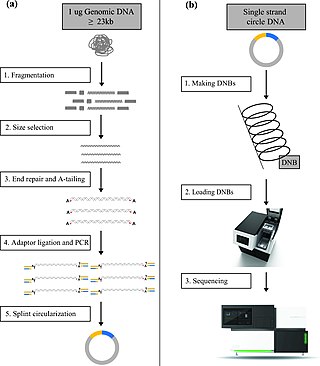
DNA nanoball sequencing is a high throughput sequencing technology that is used to determine the entire genomic sequence of an organism. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Fluorescent nucleotides bind to complementary nucleotides and are then polymerized to anchor sequences bound to known sequences on the DNA template. The base order is determined via the fluorescence of the bound nucleotides This DNA sequencing method allows large numbers of DNA nanoballs to be sequenced per run at lower reagent costs compared to other next generation sequencing platforms. However, a limitation of this method is that it generates only short sequences of DNA, which presents challenges to mapping its reads to a reference genome. After purchasing Complete Genomics, the Beijing Genomics Institute (BGI) refined DNA nanoball sequencing to sequence nucleotide samples on their own platform.
Multiple Annealing and Looping Based Amplification Cycles (MALBAC) is a quasilinear whole genome amplification method. Unlike conventional DNA amplification methods that are non-linear or exponential, MALBAC utilizes special primers that allow amplicons to have complementary ends and therefore to loop, preventing DNA from being copied exponentially. This results in amplification of only the original genomic DNA and therefore reduces amplification bias. MALBAC is “used to create overlapped shotgun amplicons covering most of the genome”. For next generation sequencing, MALBAC is followed by regular PCR which is used to further amplify amplicons.

STARR-seq is a method to assay enhancer activity for millions of candidates from arbitrary sources of DNA. It is used to identify the sequences that act as transcriptional enhancers in a direct, quantitative, and genome-wide manner.

Viral metagenomics uses metagenomic technologies to detect viral genomic material from diverse environmental and clinical samples. Viruses are the most abundant biological entity and are extremely diverse; however, only a small fraction of viruses have been sequenced and only an even smaller fraction have been isolated and cultured. Sequencing viruses can be challenging because viruses lack a universally conserved marker gene so gene-based approaches are limited. Metagenomics can be used to study and analyze unculturable viruses and has been an important tool in understanding viral diversity and abundance and in the discovery of novel viruses. For example, metagenomics methods have been used to describe viruses associated with cancerous tumors and in terrestrial ecosystems.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
Transposon insertion sequencing (Tn-seq) combines transposon insertional mutagenesis with massively parallel sequencing (MPS) of the transposon insertion sites to identify genes contributing to a function of interest in bacteria. The method was originally established by concurrent work in four laboratories under the acronyms HITS, INSeq, TraDIS, and Tn-Seq. Numerous variations have been subsequently developed and applied to diverse biological systems. Collectively, the methods are often termed Tn-Seq as they all involve monitoring the fitness of transposon insertion mutants via DNA sequencing approaches.

Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA in the bloodstream that is not associated with cells. ctDNA should not be confused with cell-free DNA (cfDNA), a broader term which describes DNA that is freely circulating in the bloodstream, but is not necessarily of tumor origin. Because ctDNA may reflect the entire tumor genome, it has gained traction for its potential clinical utility; "liquid biopsies" in the form of blood draws may be taken at various time points to monitor tumor progression throughout the treatment regimen.

Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double-stranded DNA to detect mutations with higher accuracy and lower error rates.
Ancientpathogengenomics is a scientific field related to the study of pathogen genomes recovered from ancient human, plant or animal remains. Ancient pathogens are microorganisms, now extinct, that in the past centuries caused several epidemics and deaths worldwide. Their genome, referred to as ancient DNA (aDNA), is isolated from the burial's remains of victims of the pandemics caused by these pathogens.
Clinical metagenomic next-generation sequencing (mNGS) is the comprehensive analysis of microbial and host genetic material in clinical samples from patients by next-generation sequencing. It uses the techniques of metagenomics to identify and characterize the genome of bacteria, fungi, parasites, and viruses without the need for a prior knowledge of a specific pathogen directly from clinical specimens. The capacity to detect all the potential pathogens in a sample makes metagenomic next generation sequencing a potent tool in the diagnosis of infectious disease especially when other more directed assays, such as PCR, fail. Its limitations include clinical utility, laboratory validity, sense and sensitivity, cost and regulatory considerations.
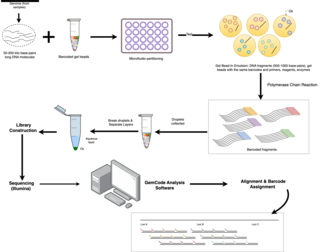
Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies.
References
- 1 2 3 4 5 6 Lan T. and Lindqvist C. 2018. Paleogenomics: Genome-Scale Analysis of Ancient DNA and Population and Evolutionary Genomic Inferences. In: Population Genomics, Springer, Cham. pp 1-38.
- 1 2 Pääbo, S. (1989-03-01). "Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification". Proceedings of the National Academy of Sciences. 86 (6): 1939–1943. Bibcode:1989PNAS...86.1939P. doi: 10.1073/pnas.86.6.1939 . ISSN 1091-6490. PMC 286820 . PMID 2928314.
- ↑ Lalueza-Fox, Carles; Castresana, Jose; Bertranpetit, Jaume; Alcover, Josep Antoni; Bover, Pere; Gigli, Elena; Ramírez, Oscar (2009-05-22). "Paleogenomics in a Temperate Environment: Shotgun Sequencing from an Extinct Mediterranean Caprine". PLOS ONE. 4 (5): e5670. Bibcode:2009PLoSO...4.5670R. doi: 10.1371/journal.pone.0005670 . ISSN 1932-6203. PMC 2680946 . PMID 19461892.
- 1 2 3 Orlando L., Gilbert MT., Willerslev E. 2015. Reconstructing ancient genomes and epigenomes. Nat. Rev. Genet. 16(7):395-408.
- ↑ Gansauge, Marie-Theres; Meyer, Matthias (September 2014). "Selective enrichment of damaged DNA molecules for ancient genome sequencing". Genome Research. 24 (9): 1543–1549. doi:10.1101/gr.174201.114. ISSN 1088-9051. PMC 4158764 . PMID 25081630.
- ↑ Carpenter, Meredith L.; Buenrostro, Jason D.; Valdiosera, Cristina; Schroeder, Hannes; Allentoft, Morten E.; Sikora, Martin; Rasmussen, Morten; Gravel, Simon; Guillén, Sonia (2013-11-07). "Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries". American Journal of Human Genetics. 93 (5): 852–864. doi:10.1016/j.ajhg.2013.10.002. ISSN 0002-9297. PMC 3824117 . PMID 24568772.
- 1 2 3 4 5 6 Skoglund P. and Mathieson I. 2018. Ancient genomics of modern humans: the first decade. Annu. Rev. Genom. Hum. Genet. 19:1, 381-404.
- 1 2 3 Marciniak S., Perry G. H. Harnessing ancient genomes to study the history of human adaptation. Nature Reviews Genetics volume 18, pages 659–674 (2017)
- ↑ Barreiro, Luis B.; Quintana-Murci, Lluís (January 2010). "From evolutionary genetics to human immunology: how selection shapes host defence genes". Nature Reviews Genetics. 11 (1): 17–30. doi: 10.1038/nrg2698 . ISSN 1471-0064. PMID 19953080. S2CID 15705508.
- ↑ Kerner, Gaspard; Neehus, Anna-Lena; Philippot, Quentin; Bohlen, Jonathan; Rinchai, Darawan; Kerrouche, Nacim; Puel, Anne; Zhang, Shen-Ying; Boisson-Dupuis, Stéphanie; Abel, Laurent; Casanova, Jean-Laurent; Patin, Etienne; Laval, Guillaume; Quintana-Murci, Lluis (8 February 2023). "Genetic adaptation to pathogens and increased risk of inflammatory disorders in post-Neolithic Europe". Cell Genomics. 3 (2): 100248. doi:10.1016/j.xgen.2022.100248. ISSN 2666-979X. PMC 9932995 . PMID 36819665. S2CID 250341156.
- ↑ Advancing the ethics of paleogenomics: Ancestral remains should not be regarded as "artifacts" but as human relatives who eserve respect - Jessica Bardill, Alyssa C. Bader, Nanibaa' A. Garrison, Deborah A. Bolnick, Jennifer A. Raff, Alexa Walker, Ripan S. Malhi, and the Summer Internship for INdigenous peoples in Genomics (SING) Consortium
| Authority control databases: National |
|---|