Related Research Articles
In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.

A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).

Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of >500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.

A Phred quality score is a measure of the quality of the identification of the nucleobases generated by automated DNA sequencing. It was originally developed for the computer program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each nucleotide base call in automated sequencer traces. The FASTQ format encodes phred scores as ASCII characters alongside the read sequences. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods. Perhaps the most important use of Phred quality scores is the automatic determination of accurate, quality-based consensus sequences.
In the fields of bioinformatics and computational biology, Genome survey sequences (GSS) are nucleotide sequences similar to expressed sequence tags (ESTs) that the only difference is that most of them are genomic in origin, rather than mRNA.
Velvet is an algorithm package that has been designed to deal with de novo genome assembly and short read sequencing alignments. This is achieved through the manipulation of de Bruijn graphs for genomic sequence assembly via the removal of errors and the simplification of repeated regions. Velvet has also been implemented in commercial packages, such as Sequencher, Geneious, MacVector and BioNumerics.
Consed is a program for viewing, editing, and finishing DNA sequence assemblies. Originally developed for sequence assemblies created with phrap, recent versions also support other sequence assembly programs like Newbler.
Phred is a computer program for base calling, that is to say, identifying a nucleobase sequence from fluorescence "trace" data generated by an automated DNA sequencer that uses electrophoresis and 4-fluorescent dye method. When originally developed, Phred produced significantly fewer errors in the data sets examined than other methods, averaging 40–50% fewer errors. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods.
The Staden Package is computer software, a set of tools for DNA sequence assembly, editing, and sequence analysis. It is open-source software, released under a BSD 3-clause license.
FASTQ format is a text-based format for storing both a biological sequence and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
SOAP is a suite of bioinformatics software tools from the BGI Bioinformatics department enabling the assembly, alignment, and analysis of next generation DNA sequencing data. It is particularly suited to short read sequencing data.

In bioinformatics, hybrid genome assembly refers to utilizing various sequencing technologies to achieve the task of assembling a genome from fragmented, sequenced DNA resulting from shotgun sequencing. Genome assembly presents one of the most challenging tasks in genome sequencing as most modern DNA sequencing technologies can only produce reads that are, on average, 25-300 base pairs in length. This is orders of magnitude smaller than the average size of a genome. This assembly is computationally difficult and has some inherent challenges, one of these challenges being that genomes often contain complex tandem repeats of sequences that can be thousands of base pairs in length. These repeats can be long enough that second generation sequencing reads are not long enough to bridge the repeat, and, as such, determining the location of each repeat in the genome can be difficult. Resolving these tandem repeats can be accomplished by utilizing long third generation sequencing reads, such as those obtained using the PacBio RS DNA sequencer. These sequences are, on average, 10,000-15,000 base pairs in length and are long enough to span most repeated regions. Using a hybrid approach to this process can increase the fidelity of assembling tandem repeats by being able to accurately place them along a linear scaffold and make the process more computationally efficient.
Base calling is the process of assigning nucleobases to chromatogram peaks or electrical current changes resulting from nucleotides passing through a nanopore. One computer program for accomplishing this job is Phred, which is a widely used base calling software program by both academic and commercial DNA sequencing laboratories because of its high base calling accuracy.
In DNA sequencing, a read is an inferred sequence of base pairs corresponding to all or part of a single DNA fragment. A typical sequencing experiment involves fragmentation of the genome into millions of molecules, which are size-selected and ligated to adapters. The set of fragments is referred to as a sequencing library, which is sequenced to produce a set of reads.
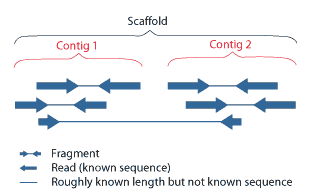
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
SPAdes is a genome assembly algorithm which was designed for single cell and multi-cells bacterial data sets. Therefore, it might not be suitable for large genomes projects.
In bioinformatics, a DNA read error occurs when a sequence assembler changes one DNA base for a different base. The reads from the sequence assembler can then be used to create a de Bruijn graph, which can be used in various ways to find errors.
De novo sequence assemblers are a type of program that assembles short nucleotide sequences into longer ones without the use of a reference genome. These are most commonly used in bioinformatic studies to assemble genomes or transcriptomes. Two common types of de novo assemblers are greedy algorithm assemblers and De Bruijn graph assemblers.
References
- ↑ Bonfield JK, Staden R (1995): The application of numerical estimates of base calling accuracy to DNA sequencing projects. Nucleic Acids Res. 1995 Apr 25;23(8):1406-10. PMID 7753633
- ↑ Krawetz SA (1989): Sequence errors described in GenBank: a means to determine the accuracy of DNA sequence interpretation. Nucleic Acids Res. 1989 May 25;17(10):3951-7