Related Research Articles
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.
Inbred strains are individuals of a particular species which are nearly identical to each other in genotype due to long inbreeding. A strain is inbred when it has undergone at least 20 generations of brother x sister or offspring x parent mating, at which point at least 98.6% of the loci in an individual of the strain will be homozygous, and each individual can be treated effectively as clones. Some inbred strains have been bred for over 150 generations, leaving individuals in the population to be isogenic in nature. Inbred strains of animals are frequently used in laboratories for experiments where for the reproducibility of conclusions all the test animals should be as similar as possible. However, for some experiments, genetic diversity in the test population may be desired. Thus outbred strains of most laboratory animals are also available, where an outbred strain is a strain of an organism that is effectively wildtype in nature, where there is as little inbreeding as possible.

A haplotype is a group of alleles in an organism that are inherited together from a single parent.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
Backcrossing is a crossing of a hybrid with one of its parents or an individual genetically similar to its parent, to achieve offspring with a genetic identity closer to that of the parent. It is used in horticulture, animal breeding, and production of gene knockout organisms.
Forward genetics is a molecular genetics approach of determining the genetic basis responsible for a phenotype. Forward genetics provides an unbiased approach because it relies heavily on identifying the genes or genetic factors that cause a particular phenotype or trait of interest.
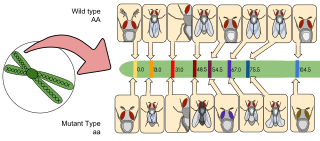
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.

In genetics, a locus is a specific, fixed position on a chromosome where a particular gene or genetic marker is located. Each chromosome carries many genes, with each gene occupying a different position or locus; in humans, the total number of protein-coding genes in a complete haploid set of 23 chromosomes is estimated at 19,000–20,000.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.
In genetics, completelinkage is defined as the state in which two loci are so close together that alleles of these loci are virtually never separated by crossing over. The closer the physical location of two genes on the DNA, the less likely they are to be separated by a crossing-over event. In the case of male Drosophila there is complete absence of recombinant types due to absence of crossing over. This means that all of the genes that start out on a single chromosome, will end up on that same chromosome in their original configuration. In the absence of recombination, only parental phenotypes are expected.
Marker assisted selection or marker aided selection (MAS) is an indirect selection process where a trait of interest is selected based on a marker linked to a trait of interest, rather than on the trait itself. This process has been extensively researched and proposed for plant- and animal- breeding.
Dr. Alexander Bachmanov studied veterinary medicine at the Saint Petersburg Veterinary Institute, Russia (1977-1982), received his Ph.D. in biological sciences from the Pavlov Institute of Physiology in Saint Petersburg, Russia in 1990. He completed postdoctoral fellowships at the Physiological Laboratory at Cambridge University in 1993 and at the Monell Chemical Senses Center, Philadelphia, Pennsylvania, in the United States from 1994 to 1997. He later joined Monnell's faculty.
A doubled haploid (DH) is a genotype formed when haploid cells undergo chromosome doubling. Artificial production of doubled haploids is important in plant breeding.
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Nested association mapping (NAM) is a technique designed by the labs of Edward Buckler, James Holland, and Michael McMullen for identifying and dissecting the genetic architecture of complex traits in corn. It is important to note that nested association mapping is a specific technique that cannot be performed outside of a specifically designed population such as the Maize NAM population, the details of which are described below.
GeneNetwork is a combined database and open-source bioinformatics data analysis software resource for systems genetics. This resource is used to study gene regulatory networks that link DNA sequence differences to corresponding differences in gene and protein expression and to variation in traits such as health and disease risk. Data sets in GeneNetwork are typically made up of large collections of genotypes and phenotypes from groups of individuals, including humans, strains of mice and rats, and organisms as diverse as Drosophila melanogaster, Arabidopsis thaliana, and barley. The inclusion of genotypes makes it practical to carry out web-based gene mapping to discover those regions of genomes that contribute to differences among individuals in mRNA, protein, and metabolite levels, as well as differences in cell function, anatomy, physiology, and behavior.
Quantitative trait loci mapping or QTL mapping is the process of identifying genomic regions that potentially contain genes responsible for important economic, health or environmental characters. Mapping QTLs is an important activity that plant breeders and geneticists routinely use to associate potential causal genes with phenotypes of interest. Family-based QTL mapping is a variant of QTL mapping where multiple-families are used.
Molecular breeding is the application of molecular biology tools, often in plant breeding and animal breeding. In the broad sense, molecular breeding can be defined as the use of genetic manipulation performed at the level of DNA to improve traits of interest in plants and animals, and it may also include genetic engineering or gene manipulation, molecular marker-assisted selection, and genomic selection. More often, however, molecular breeding implies molecular marker-assisted breeding (MAB) and is defined as the application of molecular biotechnologies, specifically molecular markers, in combination with linkage maps and genomics, to alter and improve plant or animal traits on the basis of genotypic assays.
Mega2 allows the applied statistical geneticist to convert one's data from several input formats to a large number output formats suitable for analysis by commonly used software packages. In a typical human genetics study, the analyst often needs to use a variety of different software programs to analyze the data, and these programs usually require that the data be formatted to their precise input specifications. Conversion of one's data into these multiple different formats can be tedious, time-consuming, and error-prone. Mega2, by providing validated conversion pipelines, can accelerate the analyses while reducing errors.
References
- 1 2 James F. Crow (2007). "Haldane, Bailey, Taylor and recombinant-inbred lines". Genetics . 176 (2): 729–732. doi:10.1093/genetics/176.2.729. PMC 1894602 . PMID 17579238.
- ↑ Williams RW, Gu J, Qi S, Lu L (2001). "The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis". Genome Biology . 2 (11): RESEARCH0046. doi: 10.1186/gb-2001-2-11-research0046 . PMC 59991 . PMID 11737945.
- ↑ Broman KW (2005). "The genomes of recombinant inbred lines". Genetics. 169 (2): 1133–1146. doi:10.1534/genetics.104.035212. PMC 1449115 . PMID 15545647.
- ↑ Kadarmideen HN, von Rohr P, Janss LL (2006). "From genetical genomics to systems genetics: potential applications in quantitative genomics and animal breeding". Mammalian Genome. 17 (6): 548–564. doi:10.1007/s00335-005-0169-x. PMC 3906707 . PMID 16783637.
- ↑ Morahan G, Williams RW (2007). "Systems genetics: the next generation in genetics research?". Decoding the Genomic Control of Immune Reactions. Novartis Foundation Symposia. Vol. 281. pp. 181–188. doi:10.1002/9780470062128.ch15. ISBN 9780470062128. PMID 17534074.
- ↑ Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, Magwire MM, Rollmann SM, Duncan LH, Lawrence F, Anholt RR, Mackay TF (2009). "Systems genetics of complex traits in Drosophila melanogaster". Nature Genetics . 41 (3): 299–307. doi:10.1038/ng.332. PMC 2752214 . PMID 19234471.
- ↑ Nadeau JH, Dudley AM (2011). "Genetics. Systems genetics". Science. 331 (6020): 1015–1016. doi:10.1126/science.1203869. PMC 4042627 . PMID 21350153.