Related Research Articles

Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, chemistry, physics, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.

Deoxyribonucleic acid is a polymer composed of two polynucleotide chains that coil around each other to form a double helix carrying genetic instructions for the development, functioning, growth and reproduction of all known organisms and many viruses. DNA and ribonucleic acid (RNA) are nucleic acids. Alongside proteins, lipids and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life.

Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
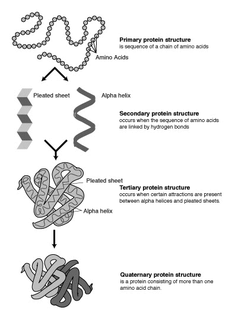
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
A protein complex or multiprotein complex is a group of two or more associated polypeptide chains. Protein complexes are distinct from multienzyme complexes, in which multiple catalytic domains are found in a single polypeptide chain.
Protein design is the rational design of new protein molecules to design novel activity, behavior, or purpose, and to advance basic understanding of protein function. Proteins can be designed from scratch or by making calculated variants of a known protein structure and its sequence. Rational protein design approaches make protein-sequence predictions that will fold to specific structures. These predicted sequences can then be validated experimentally through methods such as peptide synthesis, site-directed mutagenesis, or artificial gene synthesis.

The synaptonemal complex (SC) is a protein structure that forms between homologous chromosomes during meiosis and is thought to mediate synapsis and recombination during meiosis I in eukaryotes. It is currently thought that the SC functions primarily as a scaffold to allow interacting chromatids to complete their crossover activities.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.

Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids. It is widely used by cells to accurately repair harmful breaks that occur on both strands of DNA, known as double-strand breaks (DSB), in a process called homologous recombinational repair (HRR). Homologous recombination also produces new combinations of DNA sequences during meiosis, the process by which eukaryotes make gamete cells, like sperm and egg cells in animals. These new combinations of DNA represent genetic variation in offspring, which in turn enables populations to adapt during the course of evolution. Homologous recombination is also used in horizontal gene transfer to exchange genetic material between different strains and species of bacteria and viruses.

Directed evolution (DE) is a method used in protein engineering that mimics the process of natural selection to steer proteins or nucleic acids toward a user-defined goal. It consists of subjecting a gene to iterative rounds of mutagenesis, selection and amplification. It can be performed in vivo, or in vitro. Directed evolution is used both for protein engineering as an alternative to rationally designing modified proteins, as well as for experimental evolution studies of fundamental evolutionary principles in a controlled, laboratory environment.

DNA shuffling, also known as molecular breeding, is an in vitro random recombination method to generate mutant genes for directed evolution and to enable a rapid increase in DNA library size. Three procedures for accomplishing DNA shuffling are molecular breeding which relies on homologous recombination or the similarity of the DNA sequences, restriction enzymes which rely on common restriction sites, and nonhomologous random recombination which requires the use of hairpins. In all of these techniques, the parent genes are fragmented and then recombined.
This list of structural comparison and alignment software is a compilation of software tools and web portals used in pairwise or multiple structural comparison and structural alignment.

A protein domain is a region of the protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains. One domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
The Tn3 transposon is a 4957 base pair mobile genetic element, found in prokaryotes. It encodes three proteins:
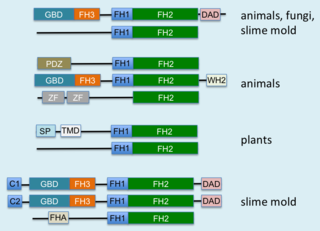
Formins are a group of proteins that are involved in the polymerization of actin and associate with the fast-growing end of actin filaments. Most formins are Rho-GTPase effector proteins. Formins regulate the actin and microtubule cytoskeleton and are involved in various cellular functions such as cell polarity, cytokinesis, cell migration and SRF transcriptional activity. Formins are multidomain proteins that interact with diverse signalling molecules and cytoskeletal proteins, although some formins have been assigned functions within the nucleus.

A circular permutation is a relationship between proteins whereby the proteins have a changed order of amino acids in their peptide sequence. The result is a protein structure with different connectivity, but overall similar three-dimensional (3D) shape. In 1979, the first pair of circularly permuted proteins – concanavalin A and lectin – were discovered; over 2000 such proteins are now known.
Infologs are independently designed synthetic genes derived from one or a few genes where substitutions are systematically incorporated to maximize information. Infologs are designed for perfect diversity distribution to maximize search efficiency.
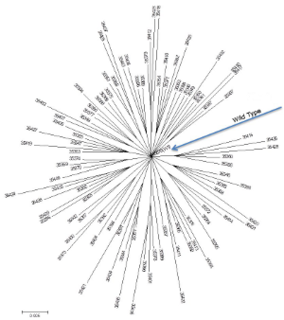
A neutral network is a set of genes all related by point mutations that have equivalent function or fitness. Each node represents a gene sequence and each line represents the mutation connecting two sequences. Neutral networks can be thought of as high, flat plateaus in a fitness landscape. During neutral evolution, genes can randomly move through neutral networks and traverse regions of sequence space which may have consequences for robustness and evolvability.
References
- ↑ Voigt, CA; Martinez, C; Wang, ZG; Mayo, SL; Arnold, FH; et al. (June 2002). "Protein building blocks preserved by recombination". Nature Structural Biology. 9 (7): 553–558. doi:10.1038/nsb805. PMID 12042875. S2CID 19170079.
- ↑ Otey, CR; Landwehr, M; Endelman, JB; Hiraga, K; Bloom, JD; Arnold, FH (May 2006). "Structure-guided recombination creates an artificial family of cytochromes P450". PLOS Biology. 4 (5): e112. doi:10.1371/journal.pbio.0040112. PMC 1431580 . PMID 16594730.

- ↑ Meyer, M; Hochrein, L.; Arnold, F (2006). "Structure-guided SCHEMA recombination of distantly related beta-lactamases". Protein Eng Des Sel. 19 (12): 563–570. doi: 10.1093/protein/gzl045 . PMID 17090554.