
Aromatic compounds or arenes usually refers to organic compounds "with a chemistry typified by benzene" and "cyclically conjugated." The word "aromatic" originates from the past grouping of molecules based on odor, before their general chemical properties were understood. The current definition of aromatic compounds does not have any relation to their odor. Aromatic compounds are now defined as cyclic compounds satisfying Hückel's Rule. Aromatic compounds have the following general properties:
In chemistry, amines are compounds and functional groups that contain a basic nitrogen atom with a lone pair. Amines are formally derivatives of ammonia, wherein one or more hydrogen atoms have been replaced by a substituent such as an alkyl or aryl group. Important amines include amino acids, biogenic amines, trimethylamine, and aniline. Inorganic derivatives of ammonia are also called amines, such as monochloramine.

In organic chemistry, hydrocarbons are divided into two classes: aromatic compounds and aliphatic compounds. Aliphatic compounds can be saturated like hexane, or unsaturated, like hexene and hexyne. Open-chain compounds, whether straight or branched, and which contain no rings of any type, are always aliphatic. Cyclic compounds can be aliphatic if they are not aromatic.
In organic chemistry, a hydrocarbon is an organic compound consisting entirely of hydrogen and carbon. Hydrocarbons are examples of group 14 hydrides. Hydrocarbons are generally colourless and hydrophobic; their odor is usually faint, and may be similar to that of gasoline or lighter fluid. They occur in a diverse range of molecular structures and phases: they can be gases, liquids, low melting solids or polymers.
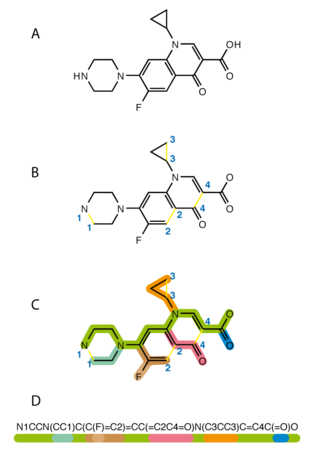
The simplified molecular-input line-entry system (SMILES) is a specification in the form of a line notation for describing the structure of chemical species using short ASCII strings. SMILES strings can be imported by most molecule editors for conversion back into two-dimensional drawings or three-dimensional models of the molecules.

In organic chemistry, the phenyl group, or phenyl ring, is a cyclic group of atoms with the formula C6H5, and is often represented by the symbol Ph. The phenyl group is closely related to benzene and can be viewed as a benzene ring, minus a hydrogen, which may be replaced by some other element or compound to serve as a functional group. A phenyl group has six carbon atoms bonded together in a hexagonal planar ring, five of which are bonded to individual hydrogen atoms, with the remaining carbon bonded to a substituent. Phenyl groups are commonplace in organic chemistry. Although often depicted with alternating double and single bonds, the phenyl group is chemically aromatic and has equal bond lengths between carbon atoms in the ring.

Kerogen is solid, insoluble organic matter in sedimentary rocks. It consists of a variety of organic materials, including dead plants, algae, and other microorganisms, that have been compressed and heated by geological processes. All the kerogen on earth is estimated to contain 1016 tons of carbon. This makes it the most abundant source of organic compounds on earth, exceeding the total organic content of living matter 10,000-fold.

In organic chemistry, aromaticity is a chemical property describing the way in which a conjugated ring of unsaturated bonds, lone pairs, or empty orbitals exhibits a stabilization stronger than would be expected by the stabilization of conjugation alone. The earliest use of the term was in an article by August Wilhelm Hofmann in 1855. There is no general relationship between aromaticity as a chemical property and the olfactory properties of such compounds.
Simple aromatic rings, also known as simple arenes or simple aromatics, are aromatic organic compounds that consist only of a conjugated planar ring system. Many simple aromatic rings have trivial names. They are usually found as substructures of more complex molecules. Typical simple aromatic compounds are benzene, indole, and pyridine.

DABCO (1,4-diazabicyclo[2.2.2]octane), also known as triethylenediamine or TEDA, is a bicyclic organic compound with the formula N2(C2H4)3. This colorless solid is a highly nucleophilic tertiary amine base, which is used as a catalyst and reagent in polymerization and organic synthesis.
In organic chemistry, Madelung synthesis is a chemical reaction that produces indoles by the intramolecular cyclization of N-phenylamides using strong base at high temperature. The Madelung synthesis was reported in 1912 by Walter Madelung, when he observed that 2-phenylindole was synthesized using N-benzoyl-o-toluidine and two equivalents of sodium ethoxide in a heated, airless reaction. Common reaction conditions include use of sodium or potassium alkoxide as base in hexane or tetrahydrofuran solvents, at temperatures ranging between 200–400 °C. A hydrolysis step is also required in the synthesis. The Madelung synthesis is important because it is one of few known reactions that produce indoles from a base-catalyzed thermal cyclization of N-acyl-o-toluidines.

Magic acid (FSO3H·SbF5) is a superacid consisting of a mixture, most commonly in a 1:1 molar ratio, of fluorosulfuric acid (HSO3F) and antimony pentafluoride (SbF5). This conjugate Brønsted–Lewis superacid system was developed in the 1960s by the George Olah lab at Case Western Reserve University, and has been used to stabilize carbocations and hypercoordinated carbonium ions in liquid media. Magic acid and other superacids are also used to catalyze isomerization of saturated hydrocarbons, and have been shown to protonate even weak bases, including methane, xenon, halogens, and molecular hydrogen.

A cyclic compound is a term for a compound in the field of chemistry in which one or more series of atoms in the compound is connected to form a ring. Rings may vary in size from three to many atoms, and include examples where all the atoms are carbon, none of the atoms are carbon, or where both carbon and non-carbon atoms are present. Depending on the ring size, the bond order of the individual links between ring atoms, and their arrangements within the rings, carbocyclic and heterocyclic compounds may be aromatic or non-aromatic; in the latter case, they may vary from being fully saturated to having varying numbers of multiple bonds between the ring atoms. Because of the tremendous diversity allowed, in combination, by the valences of common atoms and their ability to form rings, the number of possible cyclic structures, even of small size numbers in the many billions.
In medicinal chemistry, bioisosteres are chemical substituents or groups with similar physical or chemical properties which produce broadly similar biological properties in the same chemical compound. In drug design, the purpose of exchanging one bioisostere for another is to enhance the desired biological or physical properties of a compound without making significant changes in chemical structure. The main use of this term and its techniques are related to pharmaceutical sciences. Bioisosterism is used to reduce toxicity, change bioavailability, or modify the activity of the lead compound, and may alter the metabolism of the lead.
The Leuckart reaction is the chemical reaction that converts aldehydes or ketones to amines by reductive amination in the presence of heat. The reaction, named after Rudolf Leuckart, uses either ammonium formate or formamide as the nitrogen donor and reducing agent. It requires high temperatures, usually between 120 and 130 °C; for the formamide variant, the temperature can be greater than 165 °C.
The Stieglitz rearrangement is a rearrangement reaction in organic chemistry which is named after the American chemist Julius Stieglitz (1867–1937) and was first investigated by him and Paul Nicholas Leech in 1913. It describes the 1,2-rearrangement of trityl amine derivatives to triaryl imines. It is comparable to a Beckmann rearrangement which also involves a substitution at a nitrogen atom through a carbon to nitrogen shift. As an example, triaryl hydroxylamines can undergo a Stieglitz rearrangement by dehydration and the shift of a phenyl group after activation with phosphorus pentachloride to yield the respective triaryl imine, a Schiff base.
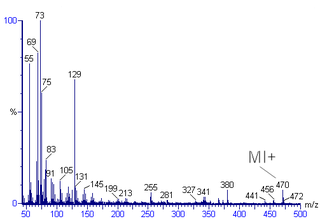
Mass spectral interpretation is the method employed to identify the chemical formula, characteristic fragment patterns and possible fragment ions from the mass spectra. Mass spectra is a plot of relative abundance against mass-to-charge ratio. It is commonly used for the identification of organic compounds from electron ionization mass spectrometry. Organic chemists obtain mass spectra of chemical compounds as part of structure elucidation and the analysis is part of many organic chemistry curricula.
Triptans is a word commonly used for a class of anti-migraine drugs that are selective 5-hydroxytryptamine/serotonin1B/1D (5-HT1B/1D) agonists. Migraine is a complex disease which affects about 15% of the population and can be highly disabling. Triptans have advantages over ergotamine and dihydroergotamine, such as selective pharmacology, well established safety record and evidence-based prescribing instructions. Triptans are therefore often preferred treatment in migraine.

An oxocarbeniumion is a chemical species characterized by a central sp2-hybridized carbon, an oxygen substituent, and an overall positive charge that is delocalized between the central carbon and oxygen atoms. An oxocarbenium ion is represented by two limiting resonance structures, one in the form of a carbenium ion with the positive charge on carbon and the other in the form of an oxonium species with the formal charge on oxygen. As a resonance hybrid, the true structure falls between the two. Compared to neutral carbonyl compounds like ketones or esters, the carbenium ion form is a larger contributor to the structure. They are common reactive intermediates in the hydrolysis of glycosidic bonds, and are a commonly used strategy for chemical glycosylation. These ions have since been proposed as reactive intermediates in a wide range of chemical transformations, and have been utilized in the total synthesis of several natural products. In addition, they commonly appear in mechanisms of enzyme-catalyzed biosynthesis and hydrolysis of carbohydrates in nature. Anthocyanins are natural flavylium dyes, which are stabilized oxocarbenium compounds. Anthocyanins are responsible for the colors of a wide variety of common flowers such as pansies and edible plants such as eggplant and blueberry.
Azidotetrazolate (CN7−) is an anion which forms a highly explosive series of salts. The ion is made by removing a proton from 5-azido-1H-tetrazole. The molecular structure contains a five-membered ring with four nitrogen atoms, and an azido side chain connected to the carbon atom. Several salts exist, but they are unstable and spontaneously explode. Rubidium azidotetrazolate was so unstable that it explodes while crystallizing. The potassium and caesium salt also spontaneously explode when dry.