Related Research Articles

Pharmacology is a branch of medicine, biology, and pharmaceutical sciences concerned with drug or medication action, where a drug may be defined as any artificial, natural, or endogenous molecule which exerts a biochemical or physiological effect on the cell, tissue, organ, or organism. It is the science of drugs including their origin, composition, pharmacokinetics, therapeutic use, and toxicology. More specifically, it is the study of the interactions that occur between a living organism and chemicals that affect normal or abnormal biochemical function. If substances have medicinal properties, they are considered pharmaceuticals.

Current good manufacturing practices (cGMP) are those conforming to the guidelines recommended by relevant agencies. Those agencies control the authorization and licensing of the manufacture and sale of food and beverages, cosmetics, pharmaceutical products, dietary supplements, and medical devices. These guidelines provide minimum requirements that a manufacturer must meet to assure that their products are consistently high in quality, from batch to batch, for their intended use. The rules that govern each industry may differ significantly; however, the main purpose of GMP is always to prevent harm from occurring to the end user. Additional tenets include ensuring the end product is free from contamination, that it is consistent in its manufacture, that its manufacture has been well documented, that personnel are well trained, and that the product has been checked for quality more than just at the end phase. GMP is typically ensured through the effective use of a quality management system (QMS).

Bioequivalence is a term in pharmacokinetics used to assess the expected in vivo biological equivalence of two proprietary preparations of a drug. If two products are said to be bioequivalent it means that they would be expected to be, for all intents and purposes, the same.
In drug development, preclinical development, also termed preclinical studies or nonclinical studies, is a stage of research that begins before clinical trials and during which important feasibility, iterative testing and drug safety data are collected, typically in laboratory animals.
Pharmacovigilance, also known as drug safety, is the pharmaceutical science relating to the "collection, detection, assessment, monitoring, and prevention" of adverse effects with pharmaceutical products. The etymological roots for the word "pharmacovigilance" are: pharmakon and vigilare. As such, pharmacovigilance heavily focuses on adverse drug reactions (ADR), which are defined as any response to a drug which is noxious and unintended, including lack of efficacy. Medication errors such as overdose, and misuse and abuse of a drug as well as drug exposure during pregnancy and breastfeeding, are also of interest, even without an adverse event, because they may result in an adverse drug reaction.

The Common Technical Document (CTD) is a set of specifications for an application dossier for the registration of Medicines and designed to be used across Europe, Japan and the United States and beyond.

hERG is a gene that codes for a protein known as Kv11.1, the alpha subunit of a potassium ion channel. This ion channel is best known for its contribution to the electrical activity of the heart: the hERG channel mediates the repolarizing IKr current in the cardiac action potential, which helps coordinate the heart's beating.
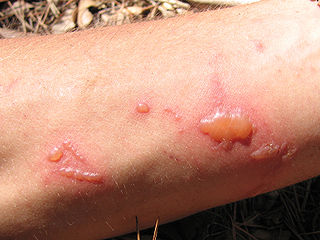
Phototoxicity, also called photoirritation, is a chemically induced skin irritation, requiring light, that does not involve the immune system. It is a type of photosensitivity.
A serious adverse event (SAE) in human drug trials is defined as any untoward medical occurrence that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage
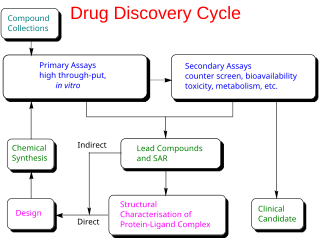
Drug development is the process of bringing a new pharmaceutical drug to the market once a lead compound has been identified through the process of drug discovery. It includes preclinical research on microorganisms and animals, filing for regulatory status, such as via the United States Food and Drug Administration for an investigational new drug to initiate clinical trials on humans, and may include the step of obtaining regulatory approval with a new drug application to market the drug. The entire process – from concept through preclinical testing in the laboratory to clinical trial development, including Phase I–III trials – to approved vaccine or drug typically takes more than a decade.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.

Bevirimat is an anti-HIV drug derived from a betulinic acid-like compound, first isolated from Syzygium claviflorum, a Chinese herb. It is believed to inhibit HIV by a novel mechanism, so-called maturation inhibition. It is not currently U.S. Food and Drug Administration (FDA) approved. It was originally developed by the pharmaceutical company Panacos and reached Phase IIb clinical trials. Myriad Genetics announced on January 21, 2009 the acquisition of all rights to bevirimat for $7M USD. On June 8, 2010 Myriad Genetics announced that it was halting the development of maturation inhibitors, including bevirimat, to focus more on their oncology portfolio.
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:

Riociguat, sold under the brand name Adempas, is a medication by Bayer that is a stimulator of soluble guanylate cyclase (sGC). It is used to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of the class of sGC stimulators. The drug has a half-life of 12 hours and will decrease dyspnea associated with pulmonary arterial hypertension.
The Safety Pharmacology Society (SPS) is a global scientific society fostering best practice around the discipline of safety pharmacology. The Society has a mission statement which declares it is a nonprofit organization that promotes knowledge, development, application, and training in safety pharmacology—a distinct scientific discipline that integrates the best practices of pharmacology, physiology and toxicology. The objective of safety pharmacology studies is to further the discovery, development and safe use of biologically active chemical entities and large molecules by the identification, monitoring and characterization of potentially undesirable pharmacodynamic activities in nonclinical studies. The Safety Pharmacology Society also supports the human safety of drugs and biologicals by fostering scientific research, education, and dissemination of scientific information through meetings and other scientific interactions.
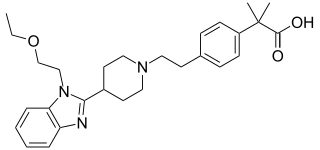
Bilastine is an antihistamine medication used to treat hives (urticaria), allergic rhinitis and itchy inflamed eyes (allergic conjunctivitis) caused by an allergy. It is a second-generation antihistamine and takes effect by selectively inhibiting the histamine H1 receptor, preventing these allergic reactions. Bilastine has an effectiveness similar to cetirizine, fexofenadine, and desloratadine.
The phases of clinical research are the stages in which scientists conduct experiments with a health intervention to obtain sufficient evidence for a process considered effective as a medical treatment. For drug development, the clinical phases start with testing for drug safety in a few human subjects, then expand to many study participants to determine if the treatment is effective. Clinical research is conducted on drug candidates, vaccine candidates, new medical devices, and new diagnostic assays.

Brexpiprazole, sold under the brand name Rexulti among others, is a medication used for the treatment of major depressive disorder, schizophrenia, and agitation associated with dementia due to Alzheimer's disease. It is an atypical antipsychotic.
Non-biological Complex Drugs (NBCDs) are medical compounds that cannot be defined as small molecular, fully identifiable drugs with active pharmaceutical ingredients. They are highly complex and cannot be defined as biologicals as they are not derived from living materials. NBCDs are synthetic complex compounds and they contain non-homomolecular, closely related molecular structures with often nanoparticular properties. This is, for instance, the case with the iron sucrose and its similars. But also with other drug products, e.g. polypeptides (glatiramoids), swelling polymers, liposomes as the NBCD class is growing. Hence and due to their complexity and specific composition mix, such colloidal iron carbohydrate drugs cannot be fully identified, characterized, quantitated and/or described by physiochemical means to define their pharmaceutical properties. Therefore, contradictory to the generic paradigm pathway, relying on a full pharmaceutical identity and sameness in vitro evaluation exercise, they need additional evaluation with a reference product to assess comparability e.g. in tissue targeting in the body. This requires an appropriate, yet to be defined and be harmonized regulatory approach for these new class of medicinal products. The profile and the performance of NBCDs is defined by the multi-step manufacturing process, which is laborious, difficult to control and not disclosed by intellectual property. Minimal changes in for instance the starting materials or the process conditions might result in significant clinical differences affecting therapeutic effects or safety.
References
- ↑ Bass A; et al. (2004). "Origins, practices and future of safety pharmacology". Journal of Pharmacological and Toxicological Methods. 49 (3): 145–151. doi:10.1016/j.vascn.2004.02.007. PMID 15172010.
- ↑ International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)
- ↑ "ICH Official web site : ICH".
- ↑ MK, Pugsley; et al. (2008). "Principles of Safety Pharmacology". Br J Pharmacol. 154 (7): 1382–1399. doi:10.1038/bjp.2008.280. PMC 2492105 . PMID 18604233.
- ↑ Sterner W, Korn WD (1980). "Zur Pharmakologie und Toxikologie von Etofyllinclofibrate". Arzneimittel-Forschung. 30 (11b): 2023–31. PMID 7194053.
- ↑ Redfern WS, Wakefield, ID (2006) Safety Pharmacology. In Toxicological Testing Handbook: Principles, Applications and Data Interpretation, 2nd edn., pp. 33-78. K Keller & D Jacobson-Kram (eds.), Taylor & Francis Ltd.
- ↑ EMA ICH S7A Safety Pharmacology Studies for Human Pharmaceuticals. ICH Step 5: Note for Guidance on Safety Pharmacology Studies for Human Pharmaceuticals. CPMP/ICH/539/00.
- ↑ Redfern WS, Wakefield ID, Prior H, Pollard, CE, Hammond TG, Valentin J-P. (2002) Safety pharmacology – a progressive approach. Fund Clin Pharmacol 16, 161-173.
- ↑ "ICH Official web site : ICH".
- ↑ Handbook of Experimental Pharmacology. Principles of Safety Pharmacology. Editors: Pugsley, Michael K., Curtis, Michael J. (Eds.), 2015.
- ↑ Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N (Mar 2014). "Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium". Am. Heart J. 167 (3): 292–300. doi: 10.1016/j.ahj.2013.11.004 . PMID 24576511.
- ↑ Authier S, Pugsley MK, Koerner JE, Fermini B, Redfern WS, Valentin JP, Vargas HM, Leishman DJ, Correll K, Curtis MJ. Proarrhythmia liability assessment and the comprehensive in vitro Proarrhythmia Assay (CiPA): An industry survey on current practice. J Pharmacol Toxicol Methods. 2017 Feb 20;86:34-43.
- ↑ Cavero; et al. (2016). "Comprehensive in vitro Proarrhythmia Assay (CiPA): Pending issues for successful validation and implementation" (PDF). Journal of Pharmacological and Toxicological Methods. 81: 21–36. doi:10.1016/j.vascn.2016.05.012. PMID 27233533. S2CID 33369081.