Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitroDNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies (like Illumina) in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%.[1] Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2[2] as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.[3]
The Sanger (chain-termination) method for DNA sequencing.
Method
Fluorescent ddNTP molecules
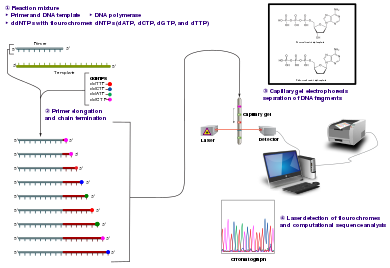
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The deoxynucleotide concentration should be approximately 100-fold higher than that of the corresponding dideoxynucleotide (e.g. 0.5mM dTTP: 0.005mM ddTTP) to allow enough fragments to be produced while still transcribing the complete sequence (but the concentration of ddNTP also depends on the desired length of sequence).[4] Putting it in a more sensible order, four separate reactions are needed in this process to test all four ddNTPs. Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. In the original publication of 1977,[4] the formation of base-paired loops of ssDNA was a cause of serious difficulty in resolving bands at some locations. This is frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image.
Part of a radioactively labelled sequencing gel
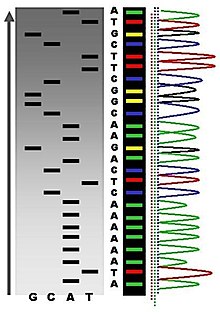
In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes, from bottom to top, are then used to read the DNA sequence.
DNA fragments are labelled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers[5][6] of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Sequence ladder by radioactive sequencing compared to fluorescent peaks
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
Dye-terminator sequencing
Capillary electrophoresis
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emits light at different wavelengths.
Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the left).
This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating "dye blobs". The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, was used for the vast majority of sequencing projects until the introduction of next generation sequencing.
Automation and sample preparation
View of the start of an example dye-terminator read
Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day. DNA sequencers separate strands by size (or length) using capillary electrophoresis, they detect and record dye fluorescence, and output data as fluorescent peak trace chromatograms. Sequencing reactions (thermocycling and labelling), cleanup and re-suspension of samples in a buffer solution are performed separately, before loading samples onto the sequencer. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (which are generally located at the ends of the sequence).[7] The accuracy of such algorithms is inferior to visual examination by a human operator, but is adequate for automated processing of large sequence data sets.
Applications of dye-terminating sequencing
The field of public health plays many roles to support patient diagnostics as well as environmental surveillance of potential toxic substances and circulating biological pathogens. Public health laboratories (PHL) and other laboratories around the world have played a pivotal role in providing rapid sequencing data for the surveillance of the virus SARS-CoV-2, causative agent for COVID-19, during the pandemic that was declared a public health emergency on January 30, 2020.[8] Laboratories were tasked with the rapid implementation of sequencing methods and asked to provide accurate data to assist in the decision-making models for the development of policies to mitigate spread of the virus. Many laboratories resorted to next generation sequencing methodologies while others supported efforts with Sanger sequencing. The sequencing efforts of SARS-CoV-2 are many, while most laboratories implemented whole genome sequencing of the virus, others have opted to sequence very specific genes of the virus such as the S-gene, encoding the information needed to produce the spike protein. The high mutation rate of SARS-CoV-2 leads to genetic differences within the S-gene and these differences have played a role in the infectivity of the virus.[9] Sanger sequencing of the S-gene provides a quick, accurate, and more affordable method to retrieving the genetic code. Laboratories in lower income countries may not have the capabilities to implement expensive applications such as next generation sequencing, so Sanger methods may prevail in supporting the generation of sequencing data for surveillance of variants.
Sanger sequencing is also the "gold standard" for norovirus surveillance methods for the Center for Disease Control and Prevention's (CDC) CaliciNet network. CalciNet is an outbreak surveillance network that was established in March 2009. The goal of the network is to collect sequencing data of circulating noroviruses in the United States and activate downstream action to determine the source of infection to mitigate the spread of the virus. The CalciNet network has identified many infections as foodborne illnesses.[3] This data can then be published and used to develop recommendations for future action to prevent tainting food. The methods employed for detection of norovirus involve targeted amplification of specific areas of the genome. The amplicons are then sequenced using dye-terminating Sanger sequencing and the chromatograms and sequences generated are analyzed with a software package developed in BioNumerics. Sequences are tracked and strain relatedness is studied to infer epidemiological relevance.
Challenges
Common challenges of DNA sequencing with the Sanger method include poor quality in the first 15-40 bases of the sequence due to primer binding and deteriorating quality of sequencing traces after 700-900 bases. Base calling software such as Phred typically provides an estimate of quality to aid in trimming of low-quality regions of sequences.[10][11]
In cases where DNA fragments are cloned before sequencing, the resulting sequence may contain parts of the cloning vector. In contrast, PCR-based cloning and next-generation sequencing technologies based on pyrosequencing often avoid using cloning vectors. Recently, one-step Sanger sequencing (combined amplification and sequencing) methods such as Ampliseq and SeqSharp have been developed that allow rapid sequencing of target genes without cloning or prior amplification.[12][13]
Current methods can directly sequence only relatively short (300-1000 nucleotides long) DNA fragments in a single reaction. The main obstacle to sequencing DNA fragments above this size limit is insufficient power of separation for resolving large DNA fragments that differ in length by only one nucleotide.
Microfluidic Sanger sequencing
Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes. This technology generates long and accurate sequence reads, while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps.
In its modern inception, high-throughput genome sequencing involves fragmenting the genome into small single-stranded pieces, followed by amplification of the fragments by polymerase chain reaction (PCR). Adopting the Sanger method, each DNA fragment is irreversibly terminated with the incorporation of a fluorescently labeled dideoxy chain-terminating nucleotide, thereby producing a DNA “ladder” of fragments that each differ in length by one base and bear a base-specific fluorescent label at the terminal base. Amplified base ladders are then separated by capillary array electrophoresis (CAE) with automated, in situ “finish-line” detection of the fluorescently labeled ssDNA fragments, which provides an ordered sequence of the fragments. These sequence reads are then computer assembled into overlapping or contiguous sequences (termed "contigs") which resemble the full genomic sequence once fully assembled.[14]
Sanger methods achieve maximum read lengths of approximately 800 bp (typically 500–600 bp with non-enriched DNA). The longer read lengths in Sanger methods display significant advantages over other sequencing methods especially in terms of sequencing repetitive regions of the genome. A challenge of short-read sequence data is particularly an issue in sequencing new genomes (de novo) and in sequencing highly rearranged genome segments, typically those seen of cancer genomes or in regions of chromosomes that exhibit structural variation.[15]
Applications of microfluidic sequencing technologies
The sequencing chip has a four-layer construction, consisting of three 100-mm-diameter glass wafers (on which device elements are microfabricated) and a polydimethylsiloxane (PDMS) membrane. Reaction chambers and capillary electrophoresis channels are etched between the top two glass wafers, which are thermally bonded. Three-dimensional channel interconnections and microvalves are formed by the PDMS and bottom manifold glass wafer.
The device consists of three functional units, each corresponding to the Sanger sequencing steps. The thermal cycling (TC) unit is a 250-nanoliter reaction chamber with integrated resistive temperature detector, microvalves, and a surface heater. Movement of reagent between the top all-glass layer and the lower glass-PDMS layer occurs through 500-μm-diameter via-holes. After thermal-cycling, the reaction mixture undergoes purification in the capture/purification chamber, and then is injected into the capillary electrophoresis (CE) chamber. The CE unit consists of a 30-cm capillary which is folded into a compact switchback pattern via 65-μm-wide turns.
Sequencing chemistry
Thermal cycling
In the TC reaction chamber, dye-terminator sequencing reagent, template DNA, and primers are loaded into the TC chamber and thermal-cycled for 35 cycles ( at 95°C for 12 seconds and at 60°C for 55 seconds).
Purification
The charged reaction mixture (containing extension fragments, template DNA, and excess sequencing reagent) is conducted through a capture/purification chamber at 30°C via a 33-Volts/cm electric field applied between capture outlet and inlet ports. The capture gel through which the sample is driven, consists of 40 μM of oligonucleotide (complementary to the primers) covalently bound to a polyacrylamide matrix. Extension fragments are immobilized by the gel matrix, and excess primer, template, free nucleotides, and salts are eluted through the capture waste port. The capture gel is heated to 67-75°C to release extension fragments.
Capillary electrophoresis
Extension fragments are injected into the CE chamber where they are electrophoresed through a 125-167-V/cm field.
Platforms
The Apollo 100 platform (Microchip Biotechnologies Inc., Dublin, CA)[16] integrates the first two Sanger sequencing steps (thermal cycling and purification) in a fully automated system. The manufacturer claims that samples are ready for capillary electrophoresis within three hours of the sample and reagents being loaded into the system. The Apollo 100 platform requires sub-microliter volumes of reagents.
Comparisons to other sequencing techniques
Performance values for genome sequencing technologies including Sanger methods and next-generation methods[15][17][18]
The ultimate goal of high-throughput sequencing is to develop systems that are low-cost, and extremely efficient at obtaining extended (longer) read lengths. Longer read lengths of each single electrophoretic separation, substantially reduces the cost associated with de novo DNA sequencing and the number of templates needed to sequence DNA contigs at a given redundancy. Microfluidics may allow for faster, cheaper and easier sequence assembly.[14]
The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.
In genetics and biochemistry, sequencing means to determine the primary structure of an unbranched biopolymer. Sequencing results in a symbolic linear depiction known as a sequence which succinctly summarizes much of the atomic-level structure of the sequenced molecule.
Southern blot is a method used for detection and quantification of a specific DNA sequence in DNA samples. This method is used in molecular biology. Briefly, purified DNA from a biological sample is digested with restriction enzymes, and the resulting DNA fragments are separated by using an electric current to move them through a sieve-like gel or matrix, which allows smaller fragments to move faster than larger fragments. The DNA fragments are transferred out of the gel or matrix onto a solid membrane, which is then exposed to a DNA probe labeled with a radioactive, fluorescent, or chemical tag. The tag allows any DNA fragments containing complementary sequences with the DNA probe sequence to be visualized within the Southern blot.
Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.
Pyrosequencing is a method of DNA sequencing based on the "sequencing by synthesis" principle, in which the sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase. Pyrosequencing relies on light detection based on a chain reaction when pyrophosphate is released. Hence, the name pyrosequencing.
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Dideoxynucleotides are chain-elongating inhibitors of DNA polymerase, used in the Sanger method for DNA sequencing. They are also known as 2',3' because both the 2' and 3' positions on the ribose lack hydroxyl groups, and are abbreviated as ddNTPs.
DNA footprinting is a method of investigating the sequence specificity of DNA-binding proteins in vitro. This technique can be used to study protein-DNA interactions both outside and within cells.
Terminal restriction fragment length polymorphism is a molecular biology technique for profiling of microbial communities based on the position of a restriction site closest to a labelled end of an amplified gene. The method is based on digesting a mixture of PCR amplified variants of a single gene using one or more restriction enzymes and detecting the size of each of the individual resulting terminal fragments using a DNA sequencer. The result is a graph image where the x-axis represents the sizes of the fragment and the y-axis represents their fluorescence intensity.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
The polymerase chain reaction (PCR) is a commonly used molecular biology tool for amplifying DNA, and various techniques for PCR optimization which have been developed by molecular biologists to improve PCR performance and minimize failure.
Single-molecule real-time (SMRT) sequencing is a parallelized single molecule DNA sequencing method. Single-molecule real-time sequencing utilizes a zero-mode waveguide (ZMW). A single DNA polymerase enzyme is affixed at the bottom of a ZMW with a single molecule of DNA as a template. The ZMW is a structure that creates an illuminated observation volume that is small enough to observe only a single nucleotide of DNA being incorporated by DNA polymerase. Each of the four DNA bases is attached to one of four different fluorescent dyes. When a nucleotide is incorporated by the DNA polymerase, the fluorescent tag is cleaved off and diffuses out of the observation area of the ZMW where its fluorescence is no longer observable. A detector detects the fluorescent signal of the nucleotide incorporation, and the base call is made according to the corresponding fluorescence of the dye.
T7 DNA polymerase is an enzyme used during the DNA replication of the T7 bacteriophage. During this process, the DNA polymerase “reads” existing DNA strands and creates two new strands that match the existing ones. The T7 DNA polymerase requires a host factor, E. coli thioredoxin, in order to carry out its function. This helps stabilize the binding of the necessary protein to the primer-template to improve processivity by more than 100-fold, which is a feature unique to this enzyme. It is a member of the Family A DNA polymerases, which include E. coli DNA polymerase I and Taq DNA polymerase.
Phred is a computer program for base calling, that is to say, identifying a nucleobase sequence from fluorescence "trace" data generated by an automated DNA sequencer that uses electrophoresis and 4-fluorescent dye method. When originally developed, Phred produced significantly fewer errors in the data sets examined than other methods, averaging 40–50% fewer errors. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods.
Optical mapping is a technique for constructing ordered, genome-wide, high-resolution restriction maps from single, stained molecules of DNA, called "optical maps". By mapping the location of restriction enzyme sites along the unknown DNA of an organism, the spectrum of resulting DNA fragments collectively serves as a unique "fingerprint" or "barcode" for that sequence. Originally developed by Dr. David C. Schwartz and his lab at NYU in the 1990s this method has since been integral to the assembly process of many large-scale sequencing projects for both microbial and eukaryotic genomes. Later technologies use DNA melting, DNA competitive binding or enzymatic labelling in order to create the optical mappings.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.
Reduced representation bisulfite sequencing (RRBS) is an efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level. It combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content. Due to the high cost and depth of sequencing to analyze methylation status in the entire genome, Meissner et al. developed this technique in 2005 to reduce the amount of nucleotides required to sequence to 1% of the genome. The fragments that comprise the reduced genome still include the majority of promoters, as well as regions such as repeated sequences that are difficult to profile using conventional bisulfite sequencing approaches.
↑ Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, etal. (1986). "Fluorescence detection in automated DNA sequence analysis". Nature. 321 (6071): 674–679. Bibcode:1986Natur.321..674S. doi:10.1038/321674a0. PMID3713851. S2CID27800972. We have developed a method for the partial automation of DNA sequence analysis. Fluorescence detection of the DNA fragments is accomplished by means of a fluorophore covalently attached to the oligonucleotide primer used in enzymatic DNA sequence analysis. A different coloured fluorophore is used for each of the reactions specific for the bases A, C, G and T. The reaction mixtures are combined and co-electrophoresed down a single polyacrylamide gel tube, the separated fluorescent bands of DNA are detected near the bottom of the tube, and the sequence information is acquired directly by computer.
Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA (October 1980). "Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing". Journal of Molecular Biology. 143 (2): 161–178. doi:10.1016/0022-2836(80)90196-5. PMID6260957.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.