
Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.

Theoretical chemistry is the branch of chemistry which develops theoretical generalizations that are part of the theoretical arsenal of modern chemistry: for example, the concepts of chemical bonding, chemical reaction, valence, the surface of potential energy, molecular orbitals, orbital interactions, and molecule activation.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
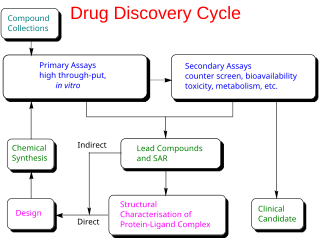
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
Quantitative structure–activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models relate a set of "predictor" variables (X) to the potency of the response variable (Y), while classification QSAR models relate the predictor variables to a categorical value of the response variable.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Amsterdam Density Functional (ADF) is a program for first-principles electronic structure calculations that makes use of density functional theory (DFT). ADF was first developed in the early seventies by the group of E. J. Baerends from the Vrije Universiteit in Amsterdam, and by the group of T. Ziegler from the University of Calgary. Nowadays many other academic groups are contributing to the software. Software for Chemistry & Materials (SCM), formerly known as Scientific Computing & Modelling is a spin-off company from the Baerends group. SCM has been coordinating the development and distribution of ADF since 1995. Together with the rise in popularity of DFT in the nineties, ADF has become a popular computational chemistry software package used in the industrial and academic research. ADF excels in spectroscopy, transition metals, and heavy elements problems. A periodic structure counterpart of ADF named BAND is available to study bulk crystals, polymers, and surfaces. The Amsterdam Modeling Suite has expanded beyond DFT since 2010, with the semi-empirical MOPAC code, the Quantum ESPRESSO plane wave code, a density-functional based tight binding (DFTB) module, a reactive force field module ReaxFF, and an implementation of Klamt's COSMO-RS method, which also includes COSMO-SAC, UNIFAC, and QSPR.

In the context of chemistry, molecular physics, physical chemistry, and molecular modelling, a force field is a computational model that is used to describe the forces between atoms within molecules or between molecules as well as in crystals. Force fields are a variety of interatomic potentials. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system on the atomistic level. Force fields are usually used in molecular dynamics or Monte Carlo simulations. The parameters for a chosen energy function may be derived from classical laboratory experiment data, calculations in quantum mechanics, or both. Force fields utilize the same concept as force fields in classical physics, with the main difference being that the force field parameters in chemistry describe the energy landscape on the atomistic level. From a force field, the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.

Chemical Computing Group is a software company specializing in research software for computational chemistry, bioinformatics, cheminformatics, docking, pharmacophore searching and molecular simulation. The company's main customer base consists of pharmaceutical and biotechnology companies, as well as academic research groups. It is a private company that was founded in 1994; it is based in Montreal, Quebec, Canada. Its main product, Molecular Operating Environment (MOE), is written in a self-contained programming system, the Scientific Vector Language (SVL).
Materials Studio is software for simulating and modeling materials. It is developed and distributed by BIOVIA, a firm specializing in research software for computational chemistry, bioinformatics, cheminformatics, molecular dynamics simulation, and quantum mechanics.
Molecular design software is notable software for molecular modeling, that provides special support for developing molecular models de novo.
Discovery Studio is a suite of software for simulating small molecule and macromolecule systems. It is developed and distributed by Dassault Systemes BIOVIA.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
LeDock is a molecular docking software, designed for protein-ligand interactions, that is compatible with Linux, macOS, and Windows.
Computational materials science and engineering uses modeling, simulation, theory, and informatics to understand materials. The main goals include discovering new materials, determining material behavior and mechanisms, explaining experiments, and exploring materials theories. It is analogous to computational chemistry and computational biology as an increasingly important subfield of materials science.