Related Research Articles

The polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation; Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, in order to distinguish individuals, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.

In genetics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.
A genetic marker is a gene or DNA sequence with a known location on a chromosome that can be used to identify individuals or species. It can be described as a variation that can be observed. A genetic marker may be a short DNA sequence, such as a sequence surrounding a single base-pair change, or a long one, like minisatellites.

AFLP-PCR or just AFLP is a PCR-based tool used in genetics research, DNA fingerprinting, and in the practice of genetic engineering. Developed in the early 1990s by KeyGene, AFLP uses restriction enzymes to digest genomic DNA, followed by ligation of adaptors to the sticky ends of the restriction fragments. A subset of the restriction fragments is then selected to be amplified. This selection is achieved by using primers complementary to the adaptor sequence, the restriction site sequence and a few nucleotides inside the restriction site fragments. The amplified fragments are separated and visualized on denaturing on agarose gel electrophoresis, either through autoradiography or fluorescence methodologies, or via automated capillary sequencing instruments.
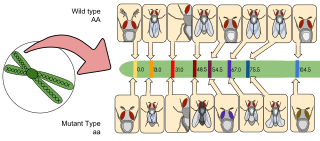
Gene mapping describes the methods used to identify the locus of a gene and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
Gene maps help describe the spatial arrangement of genes on a chromosome. Genes are designated to a specific location on a chromosome known as the locus and can be used as molecular markers to find the distance between other genes on a chromosome. Maps provide researchers with the opportunity to predict the inheritance patterns of specific traits, which can eventually lead to a better understanding of disease-linked traits.
Genotyping is the process of determining differences in the genetic make-up (genotype) of an individual by examining the individual's DNA sequence using biological assays and comparing it to another individual's sequence or a reference sequence. It reveals the alleles an individual has inherited from their parents. Traditionally genotyping is the use of DNA sequences to define biological populations by use of molecular tools. It does not usually involve defining the genes of an individual.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
An allele-specific oligonucleotide (ASO) is a short piece of synthetic DNA complementary to the sequence of a variable target DNA. It acts as a probe for the presence of the target in a Southern blot assay or, more commonly, in the simpler Dot blot assay. It is a common tool used in genetic testing, forensics, and Molecular Biology research.
In the fields of bioinformatics and computational biology, Genome survey sequences (GSS) are nucleotide sequences similar to expressed sequence tags (ESTs) that the only difference is that most of them are genomic in origin, rather than mRNA.
The following outline is provided as an overview of and topical guide to genetics:
Diversity Arrays Technology (DArT) is a high-throughput genetic marker technique that can detect allelic variations to provides comprehensive genome coverage without any DNA sequence information for genotyping and other genetic analysis. The general steps involve reducing the complexity of the genomic DNA with specific restriction enzymes, choosing diverse fragments to serve as representations for the parent genomes, amplify via polymerase chain reaction (PCR), insert fragments into a vector to be placed as probes within a microarray, then fluorescent targets from a reference sequence will be allowed to hybridize with probes and put through an imaging system. The objective is to identify and quantify various forms of DNA polymorphism within genomic DNA of sampled species.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
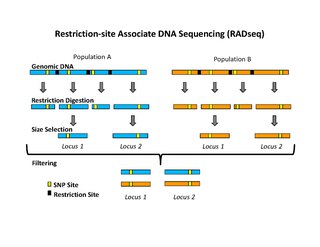
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.
Microfluidic whole genome haplotyping is a technique for the physical separation of individual chromosomes from a metaphase cell followed by direct resolution of the haplotype for each allele.
Bulked segregant analysis (BSA) is a technique used to identify genetic markers associated with a mutant phenotype. This allows geneticists to discover genes conferring certain traits of interest, such as disease resistance or susceptibility.