
Telomerase, also called terminal transferase, is a ribonucleoprotein that adds a species-dependent telomere repeat sequence to the 3' end of telomeres. A telomere is a region of repetitive sequences at each end of the chromosomes of most eukaryotes. Telomeres protect the end of the chromosome from DNA damage or from fusion with neighbouring chromosomes. The fruit fly Drosophila melanogaster lacks telomerase, but instead uses retrotransposons to maintain telomeres.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages. This can eventually lead to malignant tumors, or cancer as per the two hit hypothesis.

Non-homologous end joining (NHEJ) is a pathway that repairs double-strand breaks in DNA. NHEJ is referred to as "non-homologous" because the break ends are directly ligated without the need for a homologous template, in contrast to homology directed repair, which requires a homologous sequence to guide repair. The term "non-homologous end joining" was coined in 1996 by Moore and Haber.

Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids. It is widely used by cells to accurately repair harmful breaks that occur on both strands of DNA, known as double-strand breaks (DSB), in a process called homologous recombinational repair (HRR). Homologous recombination also produces new combinations of DNA sequences during meiosis, the process by which eukaryotes make gamete cells, like sperm and egg cells in animals. These new combinations of DNA represent genetic variation in offspring, which in turn enables populations to adapt during the course of evolution. Homologous recombination is also used in horizontal gene transfer to exchange genetic material between different strains and species of bacteria and viruses.

Dyskeratosis congenita (DKC),also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.

Ku is a dimeric protein complex that binds to DNA double-strand break ends and is required for the non-homologous end joining (NHEJ) pathway of DNA repair. Ku is evolutionarily conserved from bacteria to humans. The ancestral bacterial Ku is a homodimer. Eukaryotic Ku is a heterodimer of two polypeptides, Ku70 (XRCC6) and Ku80 (XRCC5), so named because the molecular weight of the human Ku proteins is around 70 kDa and 80 kDa. The two Ku subunits form a basket-shaped structure that threads onto the DNA end. Once bound, Ku can slide down the DNA strand, allowing more Ku molecules to thread onto the end. In higher eukaryotes, Ku forms a complex with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the full DNA-dependent protein kinase, DNA-PK. Ku is thought to function as a molecular scaffold to which other proteins involved in NHEJ can bind, orienting the double-strand break for ligation.
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are complexes of RNA and protein present in the cell nucleus during gene transcription and subsequent post-transcriptional modification of the newly synthesized RNA (pre-mRNA). The presence of the proteins bound to a pre-mRNA molecule serves as a signal that the pre-mRNA is not yet fully processed and therefore not ready for export to the cytoplasm. Since most mature RNA is exported from the nucleus relatively quickly, most RNA-binding protein in the nucleus exist as heterogeneous ribonucleoprotein particles. After splicing has occurred, the proteins remain bound to spliced introns and target them for degradation.

Serine/threonine-protein kinase ATR also known as ataxia telangiectasia and Rad3-related protein (ATR) or FRAP-related protein 1 (FRP1) is an enzyme that, in humans, is encoded by the ATR gene. It is a large kinase of about 301.66 kDa. ATR belongs to the phosphatidylinositol 3-kinase-related kinase protein family. ATR is activated in response to single strand breaks, and works with ATM to ensure genome integrity.
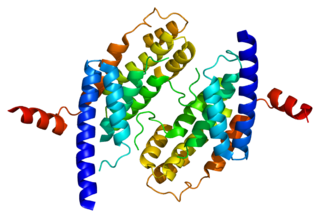
Telomeric repeat-binding factor 2 is a protein that is present at telomeres throughout the cell cycle. It is also known as TERF2, TRF2, and TRBF2, and is encoded in humans by the TERF2 gene. It is a component of the shelterin nucleoprotein complex and a second negative regulator of telomere length, playing a key role in the protective activity of telomeres. It was first reported in 1997 in the lab of Titia de Lange, where a DNA sequence similar, but not identical, to TERF1 was discovered, with respect to the Myb-domain. De Lange isolated the new Myb-containing protein sequence and called it TERF2.

Telomeric repeat-binding factor 1 is a protein that in humans is encoded by the TERF1 gene.
PIN2/TERF1-interacting telomerase inhibitor 1, also known as PINX1, is a human gene. PINX1 is also known as PIN2 interacting protein 1. PINX1 is a telomerase inhibitor and a possible tumor suppressor.
The MRN complex is a protein complex consisting of Mre11, Rad50 and Nbs1. In eukaryotes, the MRN/X complex plays an important role in the initial processing of double-strand DNA breaks prior to repair by homologous recombination or non-homologous end joining. The MRN complex binds avidly to double-strand breaks both in vitro and in vivo and may serve to tether broken ends prior to repair by non-homologous end joining or to initiate DNA end resection prior to repair by homologous recombination. The MRN complex also participates in activating the checkpoint kinase ATM in response to DNA damage. Production of short single-strand oligonucleotides by Mre11 endonuclease activity has been implicated in ATM activation by the MRN complex.

The meiotic recombination checkpoint monitors meiotic recombination during meiosis, and blocks the entry into metaphase I if recombination is not efficiently processed.
Telomere-binding proteins function to bind telomeric DNA in various species. In particular, telomere-binding protein refers to TTAGGG repeat binding factor-1 (TERF1) and TTAGGG repeat binding factor-2 (TERF2). Telomere sequences in humans are composed of TTAGGG sequences which provide protection and replication of chromosome ends to prevent degradation. Telomere-binding proteins can generate a T-loop to protect chromosome ends. TRFs are double-stranded proteins which are known to induce bending, looping, and pairing of DNA which aids in the formation of T-loops. They directly bind to TTAGGG repeat sequence in the DNA. There are also subtelomeric regions present for regulation. However, in humans, there are six subunits forming a complex known as shelterin.
Shelterin is a protein complex known to protect telomeres in many eukaryotes from DNA repair mechanisms, as well as to regulate telomerase activity. In mammals and other vertebrates, telomeric DNA consists of repeating double-stranded 5'-TTAGGG-3' (G-strand) sequences along with the 3'-AATCCC-5' (C-strand) complement, ending with a 50-400 nucleotide 3' (G-strand) overhang. Much of the final double-stranded portion of the telomere forms a T-loop (Telomere-loop) that is invaded by the 3' (G-strand) overhang to form a small D-loop (Displacement-loop).
Chromosomal instability (CIN) is a type of genomic instability in which chromosomes are unstable, such that either whole chromosomes or parts of chromosomes are duplicated or deleted. More specifically, CIN refers to the increase in rate of addition or loss of entire chromosomes or sections of them. The unequal distribution of DNA to daughter cells upon mitosis results in a failure to maintain euploidy leading to aneuploidy. In other words, the daughter cells do not have the same number of chromosomes as the cell they originated from. Chromosomal instability is the most common form of genetic instability and cause of aneuploidy.

Titia de Lange is the Director of the Anderson Center for Cancer Research, the Leon Hess professor and the head of Laboratory Cell Biology and Genetics at Rockefeller University.

Telomeric repeat-containing RNA (TERRA) is a long non-coding RNA transcribed from telomeres - repetitive nucleotide regions found on the ends of chromosomes that function to protect DNA from deterioration or fusion with neighboring chromosomes. TERRA has been shown to be ubiquitously expressed in almost all cell types containing linear chromosomes - including humans, mice, and yeasts. While the exact function of TERRA is still an active area of research, it is generally believed to play a role in regulating telomerase activity as well as maintaining the heterochromatic state at the ends of chromosomes. TERRA interaction with other associated telomeric proteins has also been shown to help regulate telomere integrity in a length-dependent manner.
Cell cycle withdrawal refers to the natural stoppage of cell cycle during cell division. When cells divide, there are many internal or external factors that would lead to a stoppage of division. This stoppage could be permanent or temporary, and could occur in any one of the four cycle phases, depending on the status of cells or the activities they are undergoing. During the process, all cell duplication process, including mitosis, meiosis as well as DNA replication, will be paused. The mechanisms involve the proteins and DNA sequences inside cells.

DNA end resection, also called 5′–3′ degradation, is a biochemical process where the blunt end of a section of double-stranded DNA is modified by cutting away some nucleotides from the 5' end to produce a 3' single-stranded sequence.
