Related Research Articles
Cheminformatics refers to the use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in structure-based drug design. The methods can also be used in chemical and allied industries, and such fields as environmental science and pharmacology, where chemical processes are involved or studied.
Quantitative structure–activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models relate a set of "predictor" variables (X) to the potency of the response variable (Y), while classification QSAR models relate the predictor variables to a categorical value of the response variable.
Mathematical chemistry is the area of research engaged in novel applications of mathematics to chemistry; it concerns itself principally with the mathematical modeling of chemical phenomena. Mathematical chemistry has also sometimes been called computer chemistry, but should not be confused with computational chemistry.
A structural analog, also known as a chemical analog or simply an analog, is a compound having a structure similar to that of another compound, but differing from it in respect to a certain component.
This page describes mining for molecules. Since molecules may be represented by molecular graphs this is strongly related to graph mining and structured data mining. The main problem is how to represent molecules while discriminating the data instances. One way to do this is chemical similarity metrics, which has a long tradition in the field of cheminformatics.
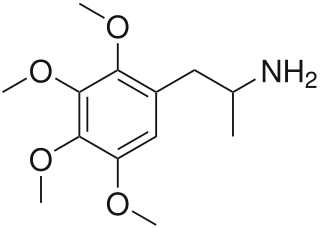
Tetramethoxyamphetamine, or 2,3,4,5-tetramethoxyamphetamine, is a lesser-known psychedelic drug and a substituted amphetamine. Tetramethoxyamphetamine was first synthesized by Alexander Shulgin. In his book PiHKAL , the minimum dosage is listed as 50 mg, and the duration unknown. Tetramethoxyamphetamine produces a threshold, mydriasis, and a headache. Limited data exists about its pharmacological properties, metabolism, and toxicity.

The Hosoya index, also known as the Z index, of a graph is the total number of matchings in it. The Hosoya index is always at least one, because the empty set of edges is counted as a matching for this purpose. Equivalently, the Hosoya index is the number of non-empty matchings plus one. The index is named after Haruo Hosoya. It is used as a topological index in chemical graph theory.
Chemical graph theory is the topology branch of mathematical chemistry which applies graph theory to mathematical modelling of chemical phenomena. The pioneers of chemical graph theory are Alexandru Balaban, Ante Graovac, Iván Gutman, Haruo Hosoya, Milan Randić and Nenad Trinajstić . In 1988, it was reported that several hundred researchers worked in this area, producing about 500 articles annually. A number of monographs have been written in the area, including the two-volume comprehensive text by Trinajstić, Chemical Graph Theory, that summarized the field up to mid-1980s.
In chemical graph theory, the Wiener index introduced by Harry Wiener, is a topological index of a molecule, defined as the sum of the lengths of the shortest paths between all pairs of vertices in the chemical graph representing the non-hydrogen atoms in the molecule.
Molecular descriptors play a fundamental role in chemistry, pharmaceutical sciences, environmental protection policy, and health researches, as well as in quality control, being the way molecules, thought of as real bodies, are transformed into numbers, allowing some mathematical treatment of the chemical information contained in the molecule. This was defined by Todeschini and Consonni as:
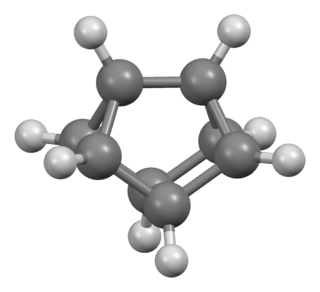
Cuneane is a saturated hydrocarbon with the formula C8H8 and a 3D structure resembling a wedge, hence the name. Cuneane may be produced from cubane by metal-ion-catalyzed σ-bond rearrangement. Similar reactions are known for homocubane and bishomocubane.

Chemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.
In chemistry, topology provides a way of describing and predicting the molecular structure within the constraints of three-dimensional (3-D) space. Given the determinants of chemical bonding and the chemical properties of the atoms, topology provides a model for explaining how the atoms ethereal wave functions must fit together. Molecular topology is a part of mathematical chemistry dealing with the algebraic description of chemical compounds so allowing a unique and easy characterization of them.

Alexandru T. Balaban is a Romanian chemist who made significant contributions to the fields of organic chemistry, theoretical chemistry, mathematical chemistry, and chemical graph theory.
Matched molecular pair analysis (MMPA) is a method in cheminformatics that compares the properties of two molecules that differ only by a single chemical transformation, such as the substitution of a hydrogen atom by a chlorine one. Such pairs of compounds are known as matched molecular pairs (MMP). Because the structural difference between the two molecules is small, any experimentally observed change in a physical or biological property between the matched molecular pair can more easily be interpreted. The term was first coined by Kenny and Sadowski in the book Chemoinformatics in Drug Discovery.
Nenad Trinajstić was a Croatian chemist and one of pioneers of the chemical graph theory.
Harry Wiener was an Austrian-American chemist, physician and psychologist, a pioneer in cheminformatics and chemical graph theory, and a long-time employee at Pfizer.
In chemical graph theory, the Padmakar–Ivan (PI) index is a topological index of a molecule, used in biochemistry. The Padmakar–Ivan index is a generalization introduced by Padmakar V. Khadikar and Iván Gutman of the concept of the Wiener index, introduced by Harry Wiener. The Padmakar–Ivan index of a graph G is the sum over all edges uv of G of number of edges which are not equidistant from u and v. Let G be a graph and e = uv an edge of G. Here denotes the number of edges lying closer to the vertex u than the vertex v, and is the number of edges lying closer to the vertex v than the vertex u. The Padmakar–Ivan index of a graph G is defined as
A chemical graph generator is a software package to generate computer representations of chemical structures adhering to certain boundary conditions. The development of such software packages is a research topic of cheminformatics. Chemical graph generators are used in areas such as virtual library generation in drug design, in molecular design with specified properties, called inverse QSAR/QSPR, as well as in organic synthesis design, retrosynthesis or in systems for computer-assisted structure elucidation (CASE). CASE systems again have regained interest for the structure elucidation of unknowns in computational metabolomics, a current area of computational biology.
The quantitative Read-Across Structure-Activity Relationship (q-RASAR) concept has been developed by merging Read-Across and QSAR. It is a statistical modeling approach that uses the similarity and error-based measures as descriptors in addition to the usual structural and physicochemical descriptors, and it has been shown to enhance the external predictivity of QSAR/QSPR models.
References
- ↑ Hendrik Timmerman; Todeschini, Roberto; Viviana Consonni; Raimund Mannhold; Hugo Kubinyi (2002). Handbook of Molecular Descriptors. Weinheim: Wiley-VCH. ISBN 3-527-29913-0.
- ↑ Hall, Lowell H.; Kier, Lemont B. (1976). Molecular connectivity in chemistry and drug research. Boston: Academic Press. ISBN 0-12-406560-0.
- ↑ González-Díaz H, Vilar S, Santana L, Uriarte E (2007). "Medicinal chemistry and bioinformatics--current trends in drugs discovery with networks topological indices". Current Topics in Medicinal Chemistry. 7 (10): 1015–29. doi:10.2174/156802607780906771. PMID 17508935.
- ↑ González-Díaz H, González-Díaz Y, Santana L, Ubeira FM, Uriarte E (February 2008). "Proteomics, networks and connectivity indices". Proteomics. 8 (4): 750–78. doi:10.1002/pmic.200700638. PMID 18297652. S2CID 20599466.
- 1 2 King, R. Bruce (1983). Chemical applications of topology and graph theory: a collection of papers from a symposium held at the University of Georgia, Athens, Georgia, U. S. A., 18–22 April 1983. Amsterdam: Elsevier. ISBN 0-444-42244-7.
- ↑ Hosoya, Haruo (1971). "Topological index. A newly proposed quantity characterizing the topological nature of structural isomers of saturated hydrocarbons". Bulletin of the Chemical Society of Japan. 44 (9): 2332–2339. doi: 10.1246/bcsj.44.2332 ..
- ↑ Katritzky AR, Karelson M, Petrukhin R (2002). "Topological Descriptors". University of Florida. Retrieved 2009-05-06.
- ↑ Pal DK, Sengupta C, De AU (1988). "A new topochemical descriptor (TAU) in molecular connectivity concept: Part I--Aliphatic compounds". Indian J. Chem. 27B: 734–739.
- ↑ Pal DK, Sengupta C, De AU (1989). "Introduction of A Novel Topochemical Index and Exploitation of Group Connectivity Concept to Achieve Predictability in QSAR and RDD". Indian J. Chem. 28B: 261–267.
- ↑ Roy K, Ghosh G (2003). "Extended Topochemical Atom (ETA) Indices in the Valence Electron Mobile (VEM) Environment as Tools". Internet Electronic Journal of Molecular Design. 2: 599–620.
- 1 2 Trofimov MI (1991). "An optimization of the procedure for the calculation of Hosoya's index". Journal of Mathematical Chemistry. 8 (1): 327–332. doi:10.1007/BF01166946. S2CID 121743373.
- ↑ Bonchev D, Mekenyan O, Trinajstić N (1981). "Isomer discrimination by topological information approach". Journal of Computational Chemistry. 2 (2): 127–148. doi:10.1002/jcc.540020202. S2CID 120705298.
- ↑ Tong W, Hong H, Xie Q, Shi L, Fang H, Perkins R (April 2005). "Assessing QSAR Limitations – A Regulatory Perspective". Current Computer-Aided Drug Design. 1 (2): 195–205. doi:10.2174/1573409053585663. Archived from the original on 2010-06-20.
- ↑ Dearden JC (2003). "In silico prediction of drug toxicity". Journal of Computer-aided Molecular Design. 17 (2–4): 119–27. Bibcode:2003JCAMD..17..119D. doi:10.1023/A:1025361621494. PMID 13677480. S2CID 21518449.
- ↑ Roy K, Ghosh G (2004). "QSTR with extended topochemical atom indices. 2. Fish toxicity of substituted benzenes". Journal of Chemical Information and Computer Sciences. 44 (2): 559–67. doi:10.1021/ci0342066. PMID 15032536.; Roy K, Ghosh G (February 2005). "QSTR with extended topochemical atom indices. Part 5: Modeling of the acute toxicity of phenylsulfonyl carboxylates to Vibrio fischeri using genetic function approximation". Bioorganic & Medicinal Chemistry. 13 (4): 1185–94. doi:10.1016/j.bmc.2004.11.014. PMID 15670927.; Roy K, Ghosh G (February 2006). "QSTR with extended topochemical atom (ETA) indices. VI. Acute toxicity of benzene derivatives to tadpoles (Rana japonica)". Journal of Molecular Modeling. 12 (3): 306–16. doi:10.1007/s00894-005-0033-7. PMID 16249936. S2CID 30293729.; Roy K, Sanyal I, Roy PP (December 2006). "QSPR of the bioconcentration factors of non-ionic organic compounds in fish using extended topochemical atom (ETA) indices". SAR and QSAR in Environmental Research. 17 (6): 563–82. Bibcode:2006SQER...17..563R. doi:10.1080/10629360601033499. PMID 17162387. S2CID 10707472.; Roy K, Ghosh G (November 2007). "QSTR with extended topochemical atom (ETA) indices. 9. Comparative QSAR for the toxicity of diverse functional organic compounds to Chlorella vulgaris using chemometric tools". Chemosphere. 70 (1): 1–12. Bibcode:2007Chmsp..70....1R. doi:10.1016/j.chemosphere.2007.07.037. PMID 17765287.